WO2011039686A1 - Latrepirdine oral sustained release dosage forms - Google Patents
Latrepirdine oral sustained release dosage forms Download PDFInfo
- Publication number
- WO2011039686A1 WO2011039686A1 PCT/IB2010/054309 IB2010054309W WO2011039686A1 WO 2011039686 A1 WO2011039686 A1 WO 2011039686A1 IB 2010054309 W IB2010054309 W IB 2010054309W WO 2011039686 A1 WO2011039686 A1 WO 2011039686A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- latrepirdine
- dosage form
- hours
- sustained release
- less
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4406—Non condensed pyridines; Hydrogenated derivatives thereof only substituted in position 3, e.g. zimeldine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0002—Galenical forms characterised by the drug release technique; Application systems commanded by energy
- A61K9/0004—Osmotic delivery systems; Sustained release driven by osmosis, thermal energy or gas
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/167—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction with an outer layer or coating comprising drug; with chemically bound drugs or non-active substances on their surface
- A61K9/1676—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction with an outer layer or coating comprising drug; with chemically bound drugs or non-active substances on their surface having a drug-free core with discrete complete coating layer containing drug
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2086—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/286—Polysaccharides, e.g. gums; Cyclodextrin
- A61K9/2866—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5005—Wall or coating material
- A61K9/5021—Organic macromolecular compounds
- A61K9/5026—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5073—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals having two or more different coatings optionally including drug-containing subcoatings
- A61K9/5078—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals having two or more different coatings optionally including drug-containing subcoatings with drug-free core
Definitions
- the invention provides sustained release formulations comprising latrepirdine or pharmaceutically acceptable salts there of.
- the formulations have desirable pharmacokinetic characteristics. Examples include AUC, C max , dosage-corrected AUC, AUC/C max , Ci2 and dosage-corrected C max .
- the present invention relates to oral sustained release compositions of 2,8- dimethyl-5-[2-(6-methylpyridin-3-yl)ethyl]-3,4-dihydro-1 H-pyrido[4,3-b]indole
- latrepirdine also known in the literature as Dimebon
- AD Alzheimer's disease
- Huntington's disease a grouping of neurodegenerative diseases, and especially Alzheimer's disease (AD) and Huntington's disease.
- Latrepirdine is being commercially developed as an immediate release tablet form with doses ranging from 5 mg to 20 mg administered TID (three times a day). While the proposed commercial dosage form provides efficacious blood levels of latrepirdine to subjects, it has been observed in clinical studies that there is a substantial amount of metabolism in most subjects. The major metabolites produced are not considered to be appreciably active relative to the parent, latrepirdine. Additionally, there is a positive food effect seen in the majority of subjects. That is, the systemic absorption of the drug is enhanced when the immediate release tablets are taken with food. It is therefore desired to provide an oral sustained release dosage form containing latrepirdine that overcomes one or more of the disadvantages of an immediate release form.
- a sustained release dosage form that decreases the dosing frequency, does not undergo the degree of metabolism as the immediate release tablet dosage form, and/or that can be taken with or without food while maintaining similar to identical bioavailability in the fed or fasted state of the subject is particularly desired.
- a sustained release formulation containing a physiologically active level of drug allows blood concentrations of the drug to be maintained at or above the therapeutic concentration. Accordingly, by achieving the sustained-release characteristics of a drug it may be possible to reduce the number of dosings while providing the same or better therapeutic effects, thus potentially improving compliance. With the sustained-release characteristics of the drug, it may also be possible to avoid a rapid increase in blood plasma concentration levels immediately after administration of the drug, thus potentially reducing or eliminating adverse side effects. There is a need in the art for new drug formulations to treat Alzheimer's disease that reduce the dosing frequency which translates to greater compliance or greater efficacy. The invention is directed to these, as well as other, important ends.
- the present invention relates to oral sustained release compositions of latrepirdine for the treatment of neurodegenerative diseases, and especially Alzheimer's disease (AD) and Huntington's disease.
- Sustained release of latrepirdine may be accomplished by any means known in the pharmaceutical arts, including but not limited to the use of osmotic dosage forms, matrix dosage forms, multiparticulate dosage forms, gastric retentive dosage forms, and pulsatile dosage forms.
- latrepirdine 2,8-dimethyl-5-[2-(6-methylpyridin-3-yl)ethyl]-3,4-dihydro-1 H-pyrido[4,3- b]indole, (hereinafter referred to as "latrepirdine”) has the following structure:
- Latrepirdine should be understood, unless otherwise indicated herein, to include any pharmaceutically acceptable form of the compound. Latrepirdine may be present in crystalline or amorphous form.
- the Amet metabolite of latrepirdine is 5-[2-(2,8-dimethyl-1 ,2,3,4-tetrahydro-5/-/- pyrido[4,3-ib]indol-5-yl)ethyl]pyridine-2-carboxylic acid, herein after referred to as "A met ", and has the following structure:
- the A met metabolite is a metabolite produced in humans when latrepirdine is orally dosed.
- a met determination in human plasma samples is completed by solid phase extraction and analyzed by liquid chromatography/tandem mass spectrometry.
- Latrepirdine is useful in the treatment of various disorders, such as Alzheimer's disease, neurodegenerative disorders, Huntington's Disease and schizophrenia (see for example, U.S. Pat. No. 6,187,785; and U.S. Pat. Appl. Pub. Nos. 2007/0117835, 2007/0117834 and 2007/0225316).
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but less than 0.1.
- the dosage form has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1.
- the dosage form has a the mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed.
- AUC 0 -inf mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed.
- C max mean maximum plasma concentration
- the C max is less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, the C max is less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a ratio of the mean area under the latrepirdine plasma concentration versus time curve (AUC 0 -i nf ) to the mean maximum latrepirdine plasma concentration (C max ) of greater than about 10.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, as a single dose in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 - in f ) of between about 0.78 ng-hr/mL per mg of latrepirdine dosed and about 1.57 ng- hr/mL per mg of latrepirdine dosed and has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.12 ng/ml per mg of latrepirdine dosed.
- AUC 0 - in f mean area under the plasma concentration versus time curve for the period following administration
- C max mean maximum plasma concentration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean latrepirdine plasma concentration twelve (12) hours post dose (d 2 ) of greater than about 0.040 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC 0 -in f ) of 0.7 to 1 .4.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose relative bioavailability test/reference crossover study, using a 20 mg latrepirdine immediate release tablet as the reference control, displays a dose normalized relative bioavailability greater than about 1 10%.
- the immediate release control tablet formulation is the Reference treatment and the sustained release formulations are the Test treatments.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
- a sustained release pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form, when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours.
- a sustained release pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form, when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours.
- a sustained release pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form, when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but less than 0.1 , and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1 .
- the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours.
- AUCo-inf plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1 . In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1 .
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A me t metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours.
- AUCo-inf plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- AUCo-inf mean area under the plasma concentration versus time curve
- AUC 0 -inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A me t metabolite greater than about 0.04 but less than 0.1.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -in f ) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours.
- AUC 0 -in f mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed.
- dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours.
- C max mean maximum plasma concentration
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours.
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed.
- dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours.
- C max mean maximum plasma concentration
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC 0 -inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC 0 -inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC 0 -i nf ) of 0.7 to 1.4, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours.
- AUC 0 -i nf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage is a sustained release form, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage is a sustained release form, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage is a sustained release form, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours.
- a sustained release pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form, when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC 0 -in f ) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
- AUC 0 -in f mean area under the plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A me t metabolite greater than about 0.04 but less than 0.1.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1 . In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1 .
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours.
- AUCo-inf mean area under the plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -i nf ) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- AUC 0 -i nf mean area under the plasma concentration versus time curve
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -in f ) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours.
- AUC 0 -in f mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed.
- dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours.
- C max mean maximum plasma concentration
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours.
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed.
- dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours.
- C max mean maximum plasma concentration
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC 0 -inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC 0 -inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours.
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC 0 -inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- AUCo-inf mean area under the plasma concentration versus time curve
- AUC 0 -inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C ma x) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
- C ma x mean maximum plasma concentration
- the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC 0 -inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1 . In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
- C max mean maximum plasma concentration
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 1 , displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 1 , displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 1 , displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours.
- a sustained release pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form, when tested in Dissolution Test 1 , displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours.
- a sustained release pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form, when tested in Dissolution Test 1 , displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours.
- a sustained release pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form, when tested in Dissolution Test 1 , displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- AUCo-inf plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1 .
- the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A me t metabolite greater than about 0.04 but less than 0.1 .
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours.
- AUCo-inf plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A me t metabolite greater than about 0.04 but less than 0.1.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUCo- inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- AUCo- inf administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -i n f) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- AUC 0 -i n f mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUCo-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours.
- AUCo-inf administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC nf ) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours.
- AUC nf mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- C max mean maximum plasma concentration
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- C max mean maximum plasma concentration
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours.
- Cmax mean maximum plasma concentration
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours.
- C max mean maximum plasma concentration
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours.
- C max mean maximum plasma concentration
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours.
- C max mean maximum plasma concentration
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours.
- C max mean maximum plasma concentration
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours.
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1 .4, and when tested in Dissolution Test 1 , displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours.
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC 0 -inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC 0 -inf) of 0.7 to 1 .4, and when tested in Dissolution Test 1 , displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC 0 -inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 2, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 2, displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 2, displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 2, displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 2, displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 2, displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- AUCo-inf plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC 0 -i n f) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours.
- AUC 0 -i n f mean area under the plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC 0 -i n f) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours.
- AUC 0 -i n f mean area under the plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1 . In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1 .
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours.
- AUCo-inf plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A me t metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours.
- AUCo-inf plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1.
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its A met metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of Latrepirdine at a rate such that 80% of the total amount of Latrepirdine is released in no less than 7 hours and no greater than 20 hours.
- AUCo-inf plasma concentration versus time curve
- the dosage form has a mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.01 but less than 0.1 . In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC 0 -inf) ratio of latrepirdine to its A met metabolite greater than about 0.04 but less than 0.1 .
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve for the period following administration
- a a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -in f ) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours.
- AUC 0 -in f mean area under the plasma concentration versus time curve for the period following administration
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- C max mean maximum plasma concentration
- the dosage form has a C ma x less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- C max mean maximum plasma concentration
- the dosage form has a C ma x less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours.
- Cmax mean maximum plasma concentration
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C ma x less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours.
- C max mean maximum plasma concentration
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours.
- C max mean maximum plasma concentration
- the dosage form has a C max less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of Latrepirdine is released in no less than 6 hours and no greater than 20 hours.
- C max mean maximum plasma concentration
- the dosage form has a C ma x less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a C max less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours.
- the C max is less than about 0.50 ng/ml per mg of latrepirdine dosed.
- the C max is less than about 0.25 ng/ml per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours.
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC 0 -inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC 0 -inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC 0 -inf) of 0.7 to 1 .4, and when tested in Dissolution Test 2, displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours.
- AUC 0 -inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours.
- AUCo-inf mean area under the plasma concentration versus time curve
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and the total daily dose of latrepirdine needed for efficacy ranges from 1mg/day to 150 mg/day.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and the total daily dose of latrepirdine needed for efficacy ranges from 5mg/day to 100 mg/day.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and the total daily dose of latrepirdine needed for efficacy ranges from 10mg/day to 75 mg/day.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and the total daily dose of latrepirdine needed for efficacy ranges from 15 mg/day to 60 mg/day.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and said latrepirdine is embedded in a matrix which releases latrepirdine by diffusion.
- a portion of the outside surface of said matrix is covered with an impermeable coating and the remainder of said outside surface is uncovered.
- this dosage form is in the form of a tablet and the uncovered surface is in the form of an opening through said impermeable coating.
- this dosage form is in the form of a tablet and the uncovered surface is in the form of a passageway which penetrates through the entire tablet.
- this dosage form is in the form of a tablet and the uncovered surface is in the form of one or more slits through said impermeable coating or in the form of one or more strips removed therefrom.
- this dosage form is substantially in the shape of a cylinder and said impermeable coating covers one or both of the opposing flat surfaces thereof. This impermeable coating may alternatively only coat the radial surface thereof.
- the matrix of this dosage form remains substantially intact during the period of latrepirdine release.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and said latrepirdine is embedded in a matrix which releases latrepirdine by eroding.
- the matrix of this dosage form comprises hydroxypropyl methylcellulose.
- the matrix of this dosage form comprises poly(ethylene oxide).
- the matrix of this dosage form comprises polyacrylic acid.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and a reservoir of latrepirdine is encased in a membrane which limits the release rate of latrepirdine by diffusion.
- this dosage form is in the form of a tablet coated with a membrane.
- a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form and is in the form of a multiparticulate comprising particles, which particles are independently coated with a membrane which limits the release rate of latrepirdine by diffusion.
- a pharmaceutical dosage form comprises a core containing latrepirdine and a semi-permeable membrane coating wherein said dosage form is a sustained release dosage form, and said coating comprises a substantially water-insoluble polymer and a solid, water-soluble polymeric material.
- said dosage form delivers latrepirdine primarily by osmotic pressure.
- the semi-permeable membrane is fabricated by an asymmetric membrane technology.
- said water insoluble polymer comprises a cellulose derivative.
- a cellulose derivative is cellulose acetate.
- the solid water soluble polymeric material comprises a polymer having an average molecular weight between 2000 and 50,000 daltons.
- the solid, water soluble polymeric material is selected from the group consisting of water soluble cellulose derivatives, acacia, dextrin, guar gum, maltodextrin, sodium alginate, starch, polyacrylates, and polyvinyl alcohols.
- Solid water soluble cellulose derivatives include but are not limited to hydroxypropylcellulose, hydroxypropylmethylcellulose and hydroxyethylcellulose.
- the core of the sustained release pharmaceutical dosage form also contains a sugar. An example of such a sugar is mannitol.
- a pharmaceutical dosage form comprises latrepirdine, a pharmaceutically acceptable carrier, and a CYP2D6 inhibitor, wherein said dosage form can be a sustained release dosage form or an immediate release dosage form.
- CYP2D6 inhibitors include but are not limited to: amiodarone, amitriptyline, bupropion, celecoxib, chlorpheniramine, chlorpromazine, cimetidine, cinacalcet, citalopram, chlorpheniramine, clomipramine, desipramine, diphenhydramine, doxepin, duloxetine, fluvoxamine, fluoxetine, goldenseal, halofantrine, haloperidol, hydroxyzine, imipramine, methadone, metoclopramide, moclobemide, paroxetine, pimozide, propafenone, quinidine/quinine, ritonavir, sertraline, terbinafine, thioridazine and
- An additional CYP2D6 inhibitor includes St. John's wort, or an extract or constituent thereof. (See Summary Of Information On Human Cyp Enzymes: Human P450 Metabolism Data by Slobodan Rendic; Drug Metabolism 34(1 &2), 83-448 (2002).)
- Some drugs and, in particular latrepirdine are metabolized by cytochrome P450 (CYP) enzymes leading to unfavorable pharmacokinetics and the need for more frequent and higher doses than are typically desirable.
- Administration of such drugs with an agent that inhibits metabolism by CYP enzymes will improve the pharmacokinetics (e.g., increase half-life, increase the time to peak plasma concentration, increase blood levels) of the drug.
- CYP cytochrome P450
- the present invention provides methods of treating neurological and psychiatric disorders comprising: administering to a mammal a pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and the amount of latrepirdine is effective for treating such disorders.
- Neurological and psychiatric disorders include but are not limited to: acute neurological and psychiatric disorders such as cerebral deficits subsequent to cardiac bypass surgery and grafting, stroke, cerebral ischemia, spinal cord trauma, head trauma, perinatal hypoxia, cardiac arrest, hypoglycemic neuronal damage, dementia, AIDS-induced dementia, vascular dementia, mixed dementias, age associated memory impairment, Alzheimer's disease, Huntington's Chorea, amyotrophic lateral sclerosis, ocular damage, retinopathy, cognitive disorders, including cognitive disorders associated with schizophrenia and bipolar disorders, idiopathic and drug- induced Parkinson's disease, muscular spasms and disorders associated with muscular spasticity including tremors, epilepsy, convulsions, migraine, migraine headache, urinary incontinence, substance tolerance, substance withdrawal, withdrawal from opiates, nicotine, tobacco products, alcohol, benzodiazepines, cocaine, sedatives, and hypnotics, psychosis, mild cognitive impairment, amnestic cognitive impairment, multi-domain cognitive
- the invention provides a method for treating a condition in a mammal, such as a human, selected from the conditions above, comprising administering a pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and the amount of latrepirdine is effective for treating such disorders, to the mammal.
- the mammal is preferably a mammal in need of such treatment.
- the invention provides a method for treating Huntington's disease, attention deficit hyperactivity disorder, schizophrenia and Alzheimer's Disease.
- the present invention provides methods of treating neurological and psychiatric disorders comprising: administering to a patient in need thereof a pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form and the amount of latrepirdine is effective for treating such disorders.
- the pharmaceutical dosage form is an immediate release dosage form and the sustained release or immediate release pharmaceutical dosage form is optionally used in combination with a CYP2D6 inhibitor to treat neurological and psychiatric disorders.
- the sustained release pharmaceutical dosage form is optionally used in combination with another active agent.
- Such an active agent may be, for example, an atypical antipsychotic, a cholinesterase inhibitor, or an NMDA receptor antagonist.
- Such atypical antipsychotics include, but are not limited to, ziprasidone, clozapine, olanzapine, risperidone, quetiapine, aripiprazole, paliperidone;
- NMDA receptor antagonists include but are not limited to memantine; and
- cholinesterase inhibitors include but are not limited to donepezil and galantamine.
- the present invention provides a method for improving the pharmacokinetics of latrepirdine comprising administering to a human in need of such treatment a therapeutically effective amount of a combination of latrepirdine and a CYP2D6 inhibitor or a pharmaceutically acceptable salt thereof.
- the present invention provides a method of sustaining latrepirdine action in a human comprising orally administering to the human a therapeutically effective amount of a combination of latrepirdine and a CYP2D6 inhibitor or a
- the present invention provides a method of treating a neurological and psychiatric disorder comprising administering to a human in need of such treatment a therapeutically effective amount of a combination of latrepirdine and a CYP2D6 inhibitor or a pharmaceutically acceptable salt thereof.
- a pharmaceutical dosage form comprises latrepirdine, a CYP2D6 inhibitor, and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC 0 -in f ) ratio of latrepirdine to its A met metabolite which is greater than 0.5 but not greater than 15.
- AUC 0 -in f mean area under the plasma concentration versus time curve
- Unit dosage forms comprising latrepirdine and a CYP2D6 inhibitor are also embraced.
- Unit dosage forms contain a predetermined dose of latrepirdine and a predetermined dose of a CYP2D6 inhibitor.
- a unit dosage form contains a predetermined dose of latrepirdine that is a therapeutically effective dose and a predetermined dose of a CYP2D6 inhibitor that reduces the rate of metabolism of latrepirdine as compared to the rate of metabolism of latrepirdine that would occur in the absence of a CYP2D6 inhibitor.
- Kits comprising latrepirdine, a CYP2D6 inhibitor and instructions for use are also provided, where the kits may contain latrepirdine and a CYP2D6 inhibitor as a unit dosage form or a separately packaged components.
- a pharmaceutical dosage form comprises latrepirdine, a CYP2D6 inhibitor, and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC 0 -in f ) of at least 10.2 ng-hr/mL per mg of latrepirdine dosed.
- a pharmaceutical dosage form comprises latrepirdine, a CYP2D6 inhibitor, and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (C max ) of latrepirdine of less than about 3 ng/mL per mg of latrepirdine dosed.
- C max mean maximum plasma concentration
- the sustained release pharmaceutical dosage forms described above can comprise latrepirdine di-hydrochloride.
- the sustained release pharmaceutical dosage forms described above can be in the form of a tablet, capsule, multiparticulate, or beads.
- any of the sustained release pharmaceutical dosage forms described can comprise a controlled release component; a delayed release and controlled release component; a delayed release and immediate release component or any combination thereof.
- the sustained release pharmaceutical dosage forms can comprise one of the following delivery technologies or a combination thereof: matrix, osmotic, reservoir, delayed release, enteric coated, or pulsatile systems.
- Sustained release dosage forms comprising latrepirdine and prepared by a method detailed herein are provided. Methods of preparing sustained release dosage forms comprising latrepirdine are also embraced.
- sustained release is meant broadly that latrepirdine is released from an oral dosage form at a rate that is slower than immediate release.
- immediate release is meant that at least 70% of the latrepirdine contained in the oral dosage form is released within 1 hour when tested in Dissolution Test 1. It is understood that oral dosage forms containing latrepirdine as the only active agent in an immediate release -only dosage form are not part of this invention.
- Oral dosage form is intended to embrace tablets, capsules, multiparticulates or beads.
- sustained release is intended to embrace an oral composition that consists of either one or a combination of the following:
- latrepirdine is interchangeable with "2,8-dimethyl-5-[2-(6- methylpyridin-3-yl)ethyl]-3,4-dihydro-1 /-/-pyrido[4,3-b]indole” and should be understood herein, unless otherwise indicated herein, to include any pharmaceutically acceptable form of the compound.
- pharmaceutically acceptable form any pharmaceutically acceptable form, including, solvates, hydrates, isomorphs, polymorphs, co-crystals, pseudomorphs, neutral forms, acid addition salt forms, and prodrugs.
- the pharmaceutically acceptable acid addition salts of latrepirdine are prepared in a conventional manner by treating a solution or suspension of the free base with about one or two chemical equivalents of a pharmaceutically acceptable acid. Conventional concentration and recrystallization techniques are employed in isolating the salts.
- Suitable acids are acetic, lactic, succinic, maleic, tartaric, citric, gluconic, ascorbic, mesylic, tosylic, benzoic, cinnamic, fumaric, sulfuric, phosphoric, hydrochloric, hydrobromic, hydroiodic, sulfamic, sulfonic such as methanesulfonic, benzenesulfonic, and related acids.
- Some preferred forms of latrepirdine include the free base and latrepirdine dihydrochloride dihydrate.
- solid oral dosage form of the present invention is a pharmaceutically- acceptable solid oral dosage form, meaning that the dosage form is safe for administration to humans and all excipients in the dosage form are pharmaceutically- acceptable, in other words safe for human ingestion.
- administered orally to a cohort of subjects having a CYP2D6 EM status in the fasted state is meant administration of latrepirdine either once (QD) or twice (BID - dosed about 12 hours apart) over a 24 hour period. Prior to the initial administration of latrepirdine, the subject has not been administered latrepirdine for a sufficient period of time (minimally 7 days) so that the subject's plasma concentration of latrepirdine is below the detectable limit.
- Cross refers to a standard testing population. Such testing populations are well known in the art and are designed to produce meaningful reproducible results. One common size is about 20 healthy subjects, equally divided between 10 males and 10 females.
- CYP2D6 EM status refers to subjects, preferably human, who have been determined by genotyping to be CYP2D6 Extensive metabolizers. They represent approximately 60-80% of the population and therefore enrolling sufficient EM subjects to run a single dose pharmacokinetic study is easily achievable. The importance of genotyping subjects is due to the variability that can be observed between poor metabolizers (PMs) and ultra metabolizers (UMs) for compounds which are metabolized extensively by CYP2D6. To identify Poor Metabolizers (PMs), Intermediate Metabolizers (IMs), Extensive Metabolizers (EMs), and Ultra Rapid Metabolizers (UMs), easy, reliable tests which require only a blood sample and less than a day for analysis are now available.
- PMs poor metabolizers
- UMs ultra metabolizers
- the AmpliChip test provides a predicted phenotype of the subject in one of the following four categories; Poor Metabolizers (PMs), Intermediate Metabolizers (IMs), Extensive Metabolizers (EMs) and Ultra Rapid Metabolizers.
- PMs Poor Metabolizers
- IMs Intermediate Metabolizers
- EMs Extensive Metabolizers
- Ultra Rapid Metabolizers Ultra Rapid Metabolizers.
- a study to measure the concentration of latrepirdine in the plasma after initial administration may be conducted using conventional methods for making such a determination.
- the study should include at least 20 subjects, preferably human, which have all been genotyped to ensure that they have CYP2D6 EM status, in order to measure mean values for C max and AUC 0 -inf-
- the study should be conducted in the fasted state.
- the subject Prior to the initial administration of latrepirdine, the subject has not been administered latrepirdine for a sufficient length of time (minimally 7 days) so that the subject's plasma concentration of latrepirdine prior to administration of the dosage form is below the detectable limit. Plasma samples are taken at a sufficient number of time points to determine C max and AUC 0 -inf-
- fasted as used herein is defined as follows: the dosing state which is defined following an overnight fast (wherein 0 caloric intake has occurred) of at least 10 hours. Subjects are administered the dosage form with 240 imL of water. No food should be allowed for at least 4 hours post-dose. Water can be allowed as desired except for one hour before and after drug administration.
- mean area under the plasma concentration versus time curve ratio of latrepirdine to its A met metabolite is meant, the individual ratio of the mean area under the plasma concentration versus time curve of latrepirdine (e.g. AUC 0 -inf) to the mean area under the plasma concentration versus time curve of its A met metabolite (e.g. AUCo-inf) is first calculated for each subject, and then the corresponding individual ratios are averaged together. In this way, it is the average of each corresponding individuals ratio which is determined.
- the latrepirdine and A met concentrations are measured by standard liquid chromatography / tandem mass spectroscopy techniques.
- the phrase "fed” as used herein is defined as follows: the dosing state which is defined following an overnight fast (wherein 0 caloric intake has occurred) of at least 10 hours, subjects then begin the recommended high fat meal 30 minutes prior to administration of the drug product. Subjects should eat this meal in 30 minutes or less; however the drug product should be administered 30 minutes after the start of the meal. The drug product should be administered with 240 imL of water. No food should be allowed for at least 4 hours post-dose. Water can be allowed as desired except for one hour before and after drug administration. A high fat (approximately 50 percent of the total caloric content of the meal is derived from fat) and high calorie (approximately 800 to 1000 calories) meal should be used as the test meal under the fed condition.
- This test meal should derive approximately 150, 250, and 500-600 calories from protein, carbohydrate, and fat respectively.
- An example test meal would be two eggs fried in butter, two strips of bacon, two slices of toast with butter, four ounces of hash brown potatoes and eight ounces of whole milk.
- the test latrepirdine solid oral dosage form can be administered to a cohort of subjects who have not eaten any food for at least ten hours and will not eat any food for at least four hours after administration of the dosage form.
- another cohort of subjects can be administered an identical latrepirdine solid oral dosage form in a fed state, for example about 30 minutes after they begin eating a United States Food and Drug Administration (FDA) standard high fat breakfast, or other meal containing a quantity of fat and calories comparable thereto, the cohort completing eating the breakfast or other meal within about 30 minutes or less of beginning the breakfast or other meal.
- FDA United States Food and Drug Administration
- the cohorts can subsequently be "switched", with the cohort which had been tested with the fasted protocol being tested according to the fed protocol, and the cohort which had been tested with the fed protocol being tested according to the fasted protocol. It is recommended that a period of time, sometimes referred to as a “washout period”, of greater than four days from completion of the first dosing pass before switching the fed or fasted protocol for each cohort.
- AUC mean area under the serum concentration versus time curve
- the individual ratio of the mean area under the plasma concentration versus time curve of latrepirdine (e.g. AUCo-inf) in the fed state to the mean area under the plasma concentration versus time curve of latrepirdine (e.g. AUC 0 -inf) in the fasted state is first calculated, and then the corresponding individual ratios are averaged together. In this way, it is the average of each corresponding individual's ratio which is determined.
- Dissolution Test 1 refers to the following test of dosage forms of latrepirdine.
- the dissolution test is conducted in a standard USP rotating paddle apparatus as disclosed in United States Pharmacopoeia (USP) Dissolution Test Chapter 711 , Apparatus 2. Paddles are rotated at 50 rpm and the dosage form is added to 500 mL of 0.2M pH 6.8 potassium phosphate buffer at 37° C. At appropriate times following test initiation (e.g., insertion of the dosage form into the apparatus), filtered aliquots (typically 1.5 mL) from the test medium are analyzed for latrepirdine by high performance liquid chromatography (HPLC). Dissolution results are reported as the percent of the total dose of latrepirdine tested dissolved versus time.
- USP United States Pharmacopoeia
- Dissolution Test 2 refers to the following test of dosage forms of latrepirdine. The test is modified from that detailed above as follows: Paddles are rotated at 100 rpm and dissolution is conducted in two stages at 37° C. Dosage forms are added to a first test medium comprising 500 mL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 mL of a dibasic and monobasic sodium phosphate buffer (total phosphate strength is 0.425M, pH 7.6), thereby converting the acid media from the first stage to a buffer having a pH of 7.5.
- a first test medium comprising 500 mL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 mL of a dibasic and monobasic sodium phosphate buffer (total phosphate strength is
- filtered aliquots typically 1.5 mL
- HPLC high performance liquid chromatography
- the individual ratio of the dose normalized mean area under the plasma concentration versus time curve of the latrepirdine sustained release test formulation (e.g. AUC 0 -inf) to the dose normalized mean area under the plasma concentration versus time curve of the latrepirdine immediate release reference formulation (e.g. AUC 0 -inf) is first calculated, and then the corresponding individual ratios are averaged together. In this way, it is the average of each corresponding individual's ratio which is determined.
- the relative bioavailability ratio of the latrepirdine Test/Reference is expressed as a simple ratio of the dose normalized mean area under the plasma concentration versus time curve of the latrepirdine test formulation (e.g. sustained release formulation) (AUC 0 -inf ng-hr/mL per mg latrepirdine)/the dose normalized mean area under the plasma concentration versus time curve of the latrepirdine reference formulation (immediate release formulation) (AUC 0 -inf ng-hr/mL per mg latrepirdine).
- latrepirdine is incorporated into an erodible or non- erodible polymeric matrix tablet.
- an erodible matrix is meant aqueous-erodible or water-swellable or aqueous-soluble in the sense of being either erodible or swellable or dissolvable in pure water or requiring the presence of an acid or base to ionize the polymeric matrix sufficiently to cause erosion or dissolution.
- the erodible polymeric matrix When contacted with the aqueous use environment, the erodible polymeric matrix imbibes water and forms an aqueous-swollen gel or "matrix" that entraps the latrepirdine.
- aqueous- swollen matrix gradually erodes, swells, disintegrates, disperses or dissolves in the environment of use, thereby controlling the release of latrepirdine to the environment of use.
- dosage forms are well known in the art. See, for example, Remington: The Science and Practice of Pharmacy, 20 th Edition, 2000.
- a key ingredient of the water-swollen matrix is the water-swellable, erodible, or soluble polymer, which may generally be described as an osmopolymer, hydrogel or water-swellable polymer.
- Such polymers may be linear, branched, or crosslinked. They may be homopolymers or copolymers.
- Exemplary polymers include naturally occurring polysaccharides such as chitin, chitosan, dextran and pullulan; gum agar, gum arabic, gum karaya, locust bean gum, gum tragacanth, carrageenans, gum ghatti, guar gum, xanthan gum and scleroglucan; starches such as dextrin and maltodextrin; hydrophilic colloids such as pectin; alginates such as ammonium alginate, sodium, potassium or calcium alginate, propylene glycol alginate; gelatin; collagen; and cellulosics.
- naturally occurring polysaccharides such as chitin, chitosan, dextran and pullulan
- gum agar gum arabic, gum karaya, locust bean gum, gum tragacanth, carrageenans, gum ghatti, guar gum, xanthan gum and scleroglucan
- starches such
- cellulosics is meant a cellulose polymer that has been modified by reaction of at least a portion of the hydroxyl groups on the saccharide repeat units with a compound to form an ester-linked or an ether-linked substituent.
- the cellulosic ethyl cellulose has an ether linked ethyl substituent attached to the saccharide repeat unit, while the cellulosic cellulose acetate has an ester linked acetate substituent.
- Cellulosics for the erodible matrix comprise aqueous-soluble and aqueous- erodible cellulosics such as ethyl cellulose (EC), methylethyl cellulose (MEC), carboxymethyl cellulose (CMC), carboxymethyl ethylcellulose (CMEC), hydroxyethyl cellulose (HEC), hydroxypropyl cellulose (HPC), cellulose acetate phthalate (CAP), cellulose acetate trimellitate (CAT), hydroxypropyl methyl cellulose (HPMC), hydroxypropyl methyl cellulose phthalate (HPMCP), hydroxypropyl methyl cellulose acetate succinate (HPMCAS), hydroxypropyl methyl cellulose acetate trimellitate (HPMCAT), and ethylhydroxy ethylcellulose (EHEC).
- EC ethyl cellulose
- MEC methylethyl cellulose
- CMC carboxymethyl cellulose
- CMEC carboxymethyl
- a particularly preferred class of such cellulosics comprises various grades of low viscosity (MW less than or equal to 50,000 daltons) and high viscosity (MW greater than 50,000 daltons) HPMC.
- Commercially available low viscosity HPMC polymers include the Dow METHOCELTM series E3, E5, E15LV, E50LV and K100LV, while high viscosity HPMC polymers include E4MCR, E10MCR, K4M, K15M and K100M; especially preferred in this group are the METHOCELTM K series.
- Other commercially available types of HPMC include the Shin Etsu METOLOSETM 90SH series.
- the HPMC has a low viscosity, meaning that the viscosity of a 2% (w/v) solution of the HPMC in water is less than about 120 cp.
- a preferred HPMC is one in which the viscosity of a 2% (w/v) solution of the HPMC in water ranges from 80 to 120 cp (such as METHOCELTM K100LV).
- erodible matrix material examples include, but are not limited to, pullulan, polyvinyl pyrrolidone, polyvinyl alcohol, polyvinyl acetate, glycerol fatty acid esters, polyacrylamide, polyacrylic acid, copolymers of ethacrylic acid or methacrylic acid (EUDRAGIT®, Rohm America, Inc., Piscataway, New Jersey) and other acrylic acid derivatives such as homopolymers and copolymers of butylmethacrylate, methylmethacrylate, ethylmethacrylate, ethylacrylate, (2-dimethylaminoethyl)methacrylate, and (trimethylaminoethyl) methacrylate chloride.
- pullulan polyvinyl pyrrolidone
- polyvinyl alcohol polyvinyl acetate
- glycerol fatty acid esters polyacrylamide
- polyacrylic acid copolymers of ethacrylic acid or me
- the erodible matrix polymer may also contain additives and excipients known in the pharmaceutical arts, including osmopolymers, osmagens, solubility-enhancing or -retarding agents and excipients that promote stability or processing of the dosage form.
- the controlled-release portion may comprise a non-erodible matrix.
- latrepirdine is distributed in an inert matrix.
- the drug is released by diffusion through the inert matrix.
- materials suitable for the inert matrix include insoluble plastics, such as copolymers of ethylene and vinyl acetate, methyl acrylate-methyl methacrylate copolymers, polyvinyl chloride, and polyethylene; hydrophilic polymers, such as ethyl cellulose, cellulose acetate, and crosslinked polyvinylpyrrolidone (also known as crospovidone); and fatty compounds, such as carnauba wax, microcrystalline wax, and triglycerides.
- insoluble plastics such as copolymers of ethylene and vinyl acetate, methyl acrylate-methyl methacrylate copolymers, polyvinyl chloride, and polyethylene
- hydrophilic polymers such as ethyl cellulose, cellulose acetate, and crosslinked polyvinylpyrroli
- a matrix multiparticulate comprises a plurality of latrepirdine-containing particles, each particle comprising a mixture of latrepirdine with one or more excipients selected to form a matrix capable of limiting the dissolution rate of the latrepirdine into an aqueous medium.
- the matrix materials useful for this embodiment are generally water-insoluble materials such as waxes, cellulose, or other water-insoluble polymers. If needed, the matrix materials may optionally be formulated with water-soluble materials which can be used as binders or as permeability-modifying agents.
- Matrix materials useful for the manufacture of these dosage forms include microcrystalline cellulose such as Avicel (registered trademark of FMC Corp., Philadelphia, Pa.), including grades of microcrystalline cellulose to which binders such as hydroxypropyl methyl cellulose have been added, waxes such as paraffin, modified vegetable oils, carnauba wax, hydrogenated castor oil, beeswax, and the like, as well as synthetic polymers such as polyvinyl chloride), polyvinyl acetate), copolymers of vinyl acetate and ethylene, polystyrene, and the like.
- microcrystalline cellulose such as Avicel (registered trademark of FMC Corp., Philadelphia, Pa.), including grades of microcrystalline cellulose to which binders such as hydroxypropyl methyl cellulose have been added, waxes such as paraffin, modified vegetable oils, carnauba wax, hydrogenated castor oil, beeswax, and the like, as well as synthetic polymers such as polyvinyl chloride), polyvinyl
- Water soluble binders or release modifying agents which can optionally be formulated into the matrix include water-soluble polymers such as hydroxypropyl cellulose (HPC), hydroxypropyl methyl cellulose (HPMC), methyl cellulose, poly (N- vinyl-2-pyrrolidinone) (PVP), poly(ethylene oxide) (PEO), poly(vinyl alcohol) (PVA), xanthan gum, carrageenan, and other such natural and synthetic materials.
- materials which function as release-modifying agents include water-soluble materials such as sugars or salts.
- Preferred water-soluble materials include lactose, sucrose, glucose, and mannitol, as well as HPC, HPMC, and PVP.
- a process for manufacturing matrix multiparticulates is the extrusion/spheronization process.
- the latrepirdine is wet-massed with a binder, extruded through a perforated plate or die, and placed on a rotating disk.
- the extrudate ideally breaks into pieces which are rounded into spheres, spheroids, or rounded rods on the rotating plate.
- Another process and composition for this method involves using water to wet-mass a blend comprising about 20 to 75% of micro-crystalline cellulose blended with, correspondingly, about 80 to 25% latrepirdine.
- Another process for manufacturing matrix multiparticulates is the preparation of wax granules.
- a desired amount of latrepirdine is stirred with liquid wax to form a homogeneous mixture, cooled and then forced through a screen to form granules.
- Preferred matrix materials are waxy substances.
- Some preferred waxy substances are hydrogenated castor oil and carnauba wax and stearyl alcohol.
- a further process for manufacturing matrix multiparticulates involves using an organic solvent to aid mixing of the latrepirdine with the matrix material.
- This technique can be used when it is desired to utilize a matrix material with an unsuitably high melting point that, if the material were employed in a molten state, would cause decomposition of the drug or of the matrix material, or would result in an unacceptable melt viscosity, thereby preventing mixing of latrepirdine with the matrix material.
- Latrepirdine and matrix material may be combined with a modest amount of solvent to form a paste, and then forced through a screen to form granules from which the solvent is then removed.
- latrepirdine and matrix material may be combined with enough solvent to completely dissolve the matrix material and the resulting solution (which may contain solid drug particles) spray dried to form the particulate dosage form.
- the matrix material is a high molecular weight synthetic polymer such as a cellulose ether or cellulose ester.
- Solvents typically employed for the process include acetone, ethanol, isopropanol, ethyl acetate, and mixtures of two or more.
- the matrix multiparticulates are formed by the melt spray congeal process.
- the melt-congeal core comprises a matrix material.
- the matrix material serves two functions. First, the matrix material allows formation of relatively smooth, round cores that are amenable to coating. Second, the matrix material binds the optional excipients and/or drugs that may be incorporated into the core.
- the matrix material has the following physical properties: a sufficiently low viscosity in the molten state to form multiparticulates, as detailed below; and rapidly congeals to a solid when cooled below its melting point.
- the matrix preferably has a melting point below that of the melting point or decomposition point of the drug, and does not substantially dissolve the drug.
- the melt-congeal cores consist essentially of a continuous phase of matrix material and optionally other excipients, with optional drug particles and optional swelling agent particles encapsulated within. Because of this, a sufficient amount of matrix material must be present to form smooth cores that are large enough to coat. In the case of cores containing solid particles, such as drug or swelling agent, the core must contain a sufficient amount of matrix material to encapsulate the drug and swelling agent to form relatively smooth and spherical cores, which are more easily coated by conventional spray-coating processes than irregularly-shaped ones.
- the matrix material may be present in the core from at least about 30 wt percent, at least about 50 wt percent, at least about 70wt percent, at least about 80 wt percent, at least about 90wt percent, and up to 100 wt percent based on the mass of the uncoated core.
- the matrix material In order to form small, smooth round cores, the matrix material must be capable of being melted and then atomized.
- the matrix material or mixture of materials is solid at 25 degrees C.
- the matrix material melts, or is capable of melting with the addition of an optional processing aid, at a temperature of less than 200 degrees centigrade so as to be suitable for melt-congeal processing described below.
- the matrix material has a melting point between 50 degrees C and 150 C C.
- the term “melt” generally refers to the transition of a crystalline material from its crystalline to its liquid state, which occurs at its melting point
- molten generally refers to such a crystalline material in its fluid state, as used herein, the terms are used more broadly.
- melt the term is used to refer to the heating of any material or mixture of materials sufficiently that it becomes fluid in the sense that it may be pumped or atomized in a manner similar to a crystalline material in the fluid state.
- molten refers to any material or mixture of materials that is in such a fluid state.
- the matrix material is selected from the group consisting of waxes, long chain alcohols (Ci 2 or greater), fatty acid esters, glycolized fatty acid esters, phosphoglycerides, polyoxyethylene alkyl ethers, long chain carboxylic acids (Ci 2 or greater), sugar alcohols, and mixtures thereof.
- Exemplary matrix materials include highly purified forms of waxes, such as Camauba wax, white and yellow beeswax, ceresin wax, microcrystalline wax, and paraffin wax; long-chain alcohols, such as stearyl alcohol, cetyl alcohol and polyethylene glycol; fatty acid esters (also known as fats or glycerides), such as isopropyl palmitate, isopropyl myristate, glyceryl monooleate, glyceryl monostearate, glyceryl palmitostearate, mixtures of mono-, di-, and trialkyl glycerides, including mixtures of glyceryl mono-, di-, and tribehenate, glyceryl tristearate, glyceryl tripalmitate and hydrogenated vegetable oils, including hydrogenated cottonseed oil; glycolized fatty acid esters, such as polyethylene glycol stearate and polyethylene glycol distearate; polyoxyethylene alkyl ethers; polyethoxylated
- the core may also contain a variety of other excipients, present in the core in an amount of from 0 to 40 wt percent, based upon the mass of the uncoated core.
- dissolution enhancer which may be used to increase the rate of water uptake by the core and consequent expansion of the swelling agent.
- the dissolution enhancer is a different material than the matrix material.
- the dissolution enhancer may be in a separate phase or a single phase with the matrix material.
- at least a portion of the dissolution enhancer is phase-separated from the matrix material.
- dissolution enhancers are amphiphilic compounds and are generally more hydrophilic than the matrix materials.
- dissolution enhancers include: surfactants such as poloxamers, docusate salts, polyoxyethylene castor oil derivatives, polysorbates, sodium lauryl sulfate, and sorbitan monoesters; sugars, such as glucose, xylitol, sorbitol and maltitol; salts, such as sodium chloride, potassium chloride, lithium chloride, calcium chloride, magnesium chloride, sodium sulfate, potassium sulfate, sodium carbonate, magnesium sulfate and potassium phosphate; and amino acids, such as alanine and glycine; and mixtures thereof.
- surfactant-type dissolution-enhancer is a poloxambetar (commercially available as the LUTROL or PLURONIC series from BASF Corp.).
- the core may also contain other optional excipients, such as agents that inhibit or delay the release of drug from the multiparticulates.
- dissolution- inhibiting agents are generally hydrophobic and include dialkylphthalates such as dibutyl phthalate, and hydrocarbon waxes, such as microcrystalline wax and paraffin wax.
- Another useful class of excipients comprises materials that may be used to adjust the viscosity of the molten feed used to form the cores. Such viscosity- adjusting excipients will generally make up 0 to 25 wt percent of the core. The viscosity of the molten feed is a key variable in obtaining cores with a narrow particle size distribution.
- the viscosity of the molten mixture be at least about 1 cp and less than about 10,000 cp, preferably at least 50 cp and less than about 1000 cp. If the molten mixture has a viscosity outside these ranges, a viscosity-adjusting agent can be added to obtain a molten mixture within the viscosity range.
- viscosity- reducing excipients include stearyl alcohol, cetyl alcohol, low molecular weight polyethylene glycol (i.e., less than about 1000 daltons), isopropyl alcohol, and water.
- viscosity-increasing excipients examples include microcrystalline wax, paraffin wax, synthetic wax, high molecular weight polyethylene glycols (i.e., greater than about 5000 daltons), ethyl cellulose, hydroxypropyl cellulose, hydroxypropyl methyl cellulose, methyl cellulose, silicon dioxide, microcrystalline cellulose, magnesium silicate, sugars, and salts.
- excipients may be added to adjust the release characteristics of the drug from the cores.
- an acid or base may be included in the composition to modify the rate at which drug is released in an aqueous use environment.
- acids or bases that can be included in the composition include citric acid, adipic acid, malic acid, fumaric acid, succinic acid, tartaric acid, di- and tribasic sodium phosphate, di- and tribasic calcium phosphate, mono-, di-, and triethanolamine, sodium bicarbonate and sodium citrate dihydrate.
- excipients may make up 0 to 25 wt percent of the core, based on the total mass of the core.
- excipients may be added to improve processing, such as excipients to reduce the static charge on the cores or to reduce the melting temperature of the matrix material.
- examples of such anti-static agents include talc and silicon dioxide.
- Flavorants, colorants, and other excipients may also be added in their usual amounts for their usual purposes. Such excipients may make up 0 to 25 wt percent of the core, based on the total mass of the core.
- the multiparticulates are made via a melt-congeal process comprising the steps: (a) forming a molten mixture comprising the drug, the glyceride and the poloxamer; (b) delivering the molten mixture of step (a) to an atomizing means to form droplets from the molten mixture; and (c) congealing the droplets from step (b) to form multiparticulates.
- the processing conditions are chosen to maintain the crystallinity of the drug.
- the temperature of the molten mixture is kept below the melting point of the drug.
- at least 70 wt percent of the drug remains crystalline within the molten feed, more preferably, at least 80 wt percent and most preferably at least 90 wt percent.
- molten mixture refers to a mixture of drug, glyceride, and poloxamer heated sufficiently that the mixture becomes sufficiently fluid that the mixture may be formed into droplets or atomized. Atomization of the molten mixture may be carried out using any of the atomization methods described below. Generally, the mixture is molten in the sense that it will flow when subjected to one or more forces such as pressure, shear, and centrifugal force, such as that exerted by a centrifugal or spinning-disk atomizer.
- the drug/glyceride/poloxamer mixture may be considered "molten" when any portion of the drug/glyceride/poloxamer mixture becomes sufficiently fluid that the mixture, as a whole, may be atomized.
- a mixture is sufficiently fluid for atomization when the viscosity of the molten mixture is less than about 20,000 cp.
- the mixture becomes molten when the mixture is heated above the melting point of the glyceride/poloxamer mixture, in cases where the glyceride/poloxamer mixture is sufficiently crystalline to have a relatively sharp melting point; or, when the glyceride/poloxamer mixture is amorphous, above the softening point of the glyceride/poloxamer mixture.
- the molten mixture is therefore often a suspension of solid particles in a fluid matrix.
- the molten mixture comprises a mixture of substantially crystalline drug particles suspended in a glyceride/poloxamer mixture that is substantially fluid. In such cases, a portion of the drug may be dissolved in the glyceride/poloxamer mixture and a portion of the glyceride/poloxamer mixture may remain solid.
- any process may be used to form the molten mixture.
- One method involves heating the glyceride/poloxamer mixture in a tank until it is fluid and then adding the drug to the molten glyceride/poloxamer mixture.
- the glyceride/poloxamer mixture is heated to a temperature of about 10 degrees C. or more above the temperature at which it becomes fluid.
- this is generally about 10 degrees C. or more above the melting point of the lowest melting point material of the mixture.
- the process is carried out so that at least a portion of the feed remains fluid until atomized.
- the drug may be added to the fluid carrier or "melt."
- the term “melt” generally refers specifically to the transition of a crystalline material from its crystalline to its liquid state, which occurs at its melting point
- the term “molten” generally refers to such a crystalline material in its fluid state, as used herein, the terms are used more broadly, referring in the case of “melt” to the heating of any material or mixture of materials sufficiently that it becomes fluid in the sense that it may be pumped or atomized in a manner similar to a crystalline material in the fluid state.
- molten refers to any material or mixture of materials that is in such a fluid state.
- the drug, the glyceride, and the poloxamer may be added to the tank and the mixture heated until the mixture has become fluid.
- the molten mixture is mixed to ensure the drug is uniformly distributed therein.
- Mixing is generally done using mechanical means, such as overhead mixers, magnetically driven mixers and stir bars, planetary mixers, and homogenizers.
- the contents of the tank can be pumped out of the tank and through an inline, static mixer or extruder and then returned to the tank.
- the amount of shear used to mix the molten feed should be sufficiently high to ensure uniform distribution of the drug in the molten carrier.
- the amount of shear is kept low enough so the form of the drug does not change, i.e., so as to cause an increase in the amount of amorphous drug or a change in the crystalline form of the drug. It is also preferred that the shear not be so high as to reduce the particle size of the drug crystals.
- the molten mixture can be mixed from a few minutes to several hours, the mixing time being dependent on the viscosity of the feed and the solubility of drug and any optional excipients in the carrier.
- An alternative method of preparing the molten mixture is to use two tanks, melting either the glyceride or the poloxamer in one tank and the other component in another tank.
- the drug is added to one of these tanks and mixed as described above.
- the two melts are then pumped through an in-line static mixer or extruder to produce a single molten mixture that is directed to the atomization process described below.
- Another method that can be used to prepare the molten mixture is to use a continuously stirred tank system.
- the drug, glyceride, and poloxamer are continuously added to a heated tank equipped with means for continuous stirring, while the molten feed is continuously removed from the tank.
- the contents of the tank are heated such that the temperature of the contents is about 10 degrees C. or more above the melting point of the carrier.
- the drug, glyceride, and poloxamer are added in such proportions that the molten mixture removed from the tank has the desired composition.
- the drug is typically added in solid form and may be pre-heated prior to addition to the tank.
- the glyceride and poloxamer may also be preheated or even pre-melted prior to addition to the continuously stirred tank system.
- extruder In another method for forming the molten mixture is by an extruder.
- extruder is meant a device or collection of devices that creates a molten extrudate by heat and/or shear forces and/or produces a uniformly mixed extrudate from a solid and/or liquid (e.g., molten) feed.
- Such devices include, but are not limited to single- screw extruders; twin-screw extruders, including co-rotating, counter-rotating, intermeshing, and non-intermeshing extruders; multiple screw extruders; ram extruders, consisting of a heated cylinder and a piston for extruding the molten feed; gear-pump extruders, consisting of a heated gear pump, generally counter-rotating, that simultaneously heats and pumps the molten feed; and conveyer extruders.
- Conveyer extruders comprise a conveyer means for transporting solid and/or powdered feeds, such, such as a screw conveyer or pneumatic conveyer, and a pump.
- At least a portion of the conveyer means is heated to a sufficiently high temperature to produce the molten mixture.
- the molten mixture may optionally be directed to an accumulation tank, before being directed to a pump, which directs the molten mixture to an atomizer.
- an in-line mixer may be used before or after the pump to ensure the molten mixture is substantially homogeneous.
- the molten mixture is mixed to form a uniformly mixed extrudate.
- Such mixing may be accomplished by various mechanical and processing means, including mixing elements, kneading elements, and shear mixing by backflow.
- the composition is fed to the extruder, which produces a molten mixture that can be directed to the atomizer.
- the composition is fed to the extruder in the form of a solid powder.
- the powdered feed can be prepared using methods well known in the art for obtaining powdered mixtures with high content uniformity. Generally, it is desirable that the particle sizes of the drug, glyceride, and poloxamer be similar to obtain a substantially uniform blend. However, this is not essential to the successful practice of the invention.
- An example of a process for preparing a substantially uniform blend is as follows. First, the glyceride and poloxamer are milled so that their particle sizes are about the same as that of the drug; next, the drug, glyceride, and poloxamer are blended in a V-blender for 20 minutes; the resulting blend is then de-lumped to remove large particles; the resulting blend is finally blended for an additional 4 minutes. In some cases it is difficult to mill the glyceride and poloxamer to the desired particle size since many of these materials tend to be waxy substances and the heat generated during the milling process can gum up the milling equipment.
- small particles of the glyceride and poloxamer can be formed using a melt- or spray-congeal process, as described below.
- the resulting congealed particles of glyceride and poloxamer can then be blended with the drug to produce the feed for the extruder.
- Another method for producing the feed to the extruder is to melt the glyceride and poloxamer in a tank, mix in the drug as described above for the tank system, and then cool the molten mixture, producing a solidified mixture of drug and carrier. This solidified mixture can then be milled to a uniform particle size and fed to the extruder.
- a two-feed extruder system can also be used to produce the molten mixture. In this system the drug, glyceride, and poloxamer, all in powdered form, are fed to the extruder through the same or different feed ports. In this way, the need for blending the components is eliminated.
- the glyceride and poloxamer in powder form may be fed to the extruder at one point, allowing the extruder to melt the glyceride and poloxamer.
- the drug is then added to the molten glyceride and poloxamer through a second feed delivery port part way along the length of the extruder, thus minimizing the contact time of the drug with the molten glyceride and poloxamer.
- Multiple-feed extruders can be used when optional excipients are included in the multiparticulate.
- the composition is in the form of large solid particles or a solid mass, rather than a powder, when fed to the extruder.
- a solidified mixture can be prepared as described above and then molded to fit into the cylinder of a ram extruder and used directly without milling.
- the glyceride and poloxamer can be first melted in, for example, a tank, and fed to the extruder in molten form.
- the drug typically in powdered form, may then be introduced to the extruder through the same or a different delivery port used to feed the glyceride and poloxamer into the extruder.
- This system has the advantage of separating the melting step for the glyceride and poloxamer from the mixing step, minimizing contact of the drug with the molten glyceride and poloxamer.
- the extruder should be designed such that it produces a molten mixture with the drug crystals uniformly distributed in the glyceride/poloxamer mixture.
- the temperature of the extrudate should be about 10 degrees C. or more above the temperature at which the drug and carrier mixture becomes fluid.
- the various zones in the extruder should be heated to appropriate temperatures to obtain the desired extrudate temperature as well as the desired degree of mixing or shear, using procedures well known in the art. As discussed above for mechanical mixing, a minimum shear should be used to produce a uniform molten mixture, such that the crystalline form of the drug is unchanged and that dissolution or formation of amorphous drug is minimized.
- the feed is preferably molten prior to congealing for at least 5 seconds, more preferably at least 10 seconds, and most preferably at least 15 seconds, so as to ensure adequate homogeneity of the drug/glyceride/poloxamer melt. It is also preferred that the molten mixture remain molten for no more than about 20 minutes to limit exposure of the drug to the molten mixture. As described above, depending on the reactivity of the chosen glyceride/poloxamer mixture, it may be preferable to further reduce the time that the mixture is molten to well below 20 minutes in order to limit drug degradation to an acceptable level. In such cases, such mixtures may be maintained in the molten state for less than 15 minutes, and in some cases, even less than 10 minutes.
- the times above refer to the mean time from when material is introduced to the extruder to when the molten mixture is congealed. Such mean times can be determined by procedures well known in the art.
- a small amount of dye or other similar compound is added to the feed while the extruder is operating under nominal conditions. Congealed multiparticulates are then collected over time and analyzed for the dye, from which the mean time is determined.
- the drug when the drug is a crystalline hydrate, it may be desirable to maintain a high water activity in the drug/glyceride/poloxamer admixture to reduce dehydration of the drug. This can be accomplished either by adding water to the powdered feed blend or by injecting water directly into the extruder by metering a controlled amount of water into a separate delivery port. In either case, sufficient water should be added to ensure the water activity is high enough to maintain the desired form of the crystalline drug. Generally, it is desirable to keep the water activity of any material in contact with drug hydrate in the 30 percent to 100 percent RH range.
- the molten mixture is delivered to an atomizer that breaks the molten feed into small droplets.
- Virtually any method can be used to deliver the molten mixture to the atomizer, including the use of pumps and various types of pneumatic devices (e.g., pressurized vessels, piston pots).
- the extruder itself can be used to deliver the molten mixture to the atomizer.
- the molten mixture is maintained at an elevated temperature while delivering the mixture to the atomizer to prevent solidification of the mixture and to keep the molten mixture flowing.
- atomization occurs in one of several ways, including (1 ) by
- pressure or single-fluid nozzles; (2) by two-fluid nozzles; (3) by centrifugal or spinning-disk atomizers, (4) by ultrasonic nozzles; and (5) by mechanical vibrating nozzles.
- a centrifugal or spinning-disk atomizer is used, such as the FX1 100-mm rotary atomizer manufactured by Niro A S (Soeborg, Denmark).
- the droplets are congealed, typically by contact with a gas or liquid at a temperature below the solidification temperature of the droplets. Typically, it is desirable that the droplets are congealed in less than about 60 seconds, preferably in less than about 10 seconds, more preferably in less than about 1 second. Often, congealing at ambient temperature results in sufficiently rapid solidification of the droplets. However, the congealing step often occurs in an enclosed space to simplify collection of the multiparticulates. In such cases, the temperature of the congealing media (either gas or liquid) will increase over time as the droplets are introduced into the enclosed space, potentially effecting the formation of the multiparticulates or the chemical stability of the drug.
- a cooling gas or liquid is often circulated through the enclosed space to maintain a constant congealing temperature.
- the cooling gas or liquid can be cooled to below ambient temperature to promote rapid congealing, thus minimizing formation of degradants.
- the multiparticulates Following formation of the multiparticulates, it may be desired to post-treat the multiparticulates to improve drug crystallinity and/or the stability of the multiparticulate.
- the multiparticulates may also be mixed or blended with one or more pharmaceutically acceptable materials to form a suitable dosage form.
- suitable dosage forms include tablets, capsules, sachets, oral powders for constitution, and the like.
- the multiparticulates may optionally be coated with an additional exterior coating.
- the exterior coating may be any conventional coating, such as a protective film coating, a coating to provide delayed or sustained release of the drug, or to provide tastemasking.
- the coating is an enteric coating to provide delayed release of the drug.
- enteric coating is meant an acid resistant coating that remains intact and does not dissolve at pH of less than about 4.
- the enteric coating surrounds the multiparticulate so that the solid amorphous dispersion layer does not dissolve or erode in the stomach.
- the enteric coating may include an enteric coating polymer.
- Enteric coating polymers are generally polyacids having a pK a of about 3 to 5.
- enteric coating polymers include: cellulose derivatives, such as cellulose acetate phthalate, cellulose acetate trimellitate, hydroxypropyl methyl cellulose acetate succinate, cellulose acetate succinate, carboxy methyl ethyl cellulose, methylcellulose phthalate, and ethylhydroxy cellulose phthalate; vinyl polymers, such as polyvinyl acetate phthalate, vinyl acetate-maleic anhydride copolymer; polyacrylates; and polymethacrylates such as methyl acrylate-methacrylic acid copolymer, methacrylate-methacrylic acid-octyl acrylate copolymer; and styrene- maleic mono-ester copolymer.
- cellulose derivatives such as cellulose acetate phthalate, cellulose acetate trimellitate, hydroxypropyl methyl cellulose acetate succinate, cellulose acetate succinate, carboxy methyl ethyl cellulose, methylcellulose
- enteric coating materials are the pharmaceutically acceptable methacrylic acid copolymer which are copolymers, anionic in character, based on methacrylic acid and methyl methacrylate. Some of these polymers are known and sold as enteric polymers, for example having a solubility in aqueous media at pH 5.5 and above, such as the commercially available EUDRAGIT enteric polymers, such as Eudragit L 30, a polymer synthesized from dimethylaminoethyl methacrylate and Eudragit S and Eudragit FS.
- EUDRAGIT enteric polymers such as Eudragit L 30, a polymer synthesized from dimethylaminoethyl methacrylate and Eudragit S and Eudragit FS.
- the exterior coatings may include conventional plasticizers, including dibutyl phthalate; dibutyl sebacate; diethyl phthalate; dimethyl phthalate; triethyl citrate; benzyl benzoate; butyl and glycol esters of fatty acids; mineral oil; oleic acid; stearic acid; cetyl alcohol; stearyl alcohol; castor oil; corn oil; coconut oil; and camphor oil; and other excipients such as anti-tack agents, glidants, etc.
- plasticizers triethyl citrate, coconut oil and dibutyl sebacate are particularly preferred.
- Exterior coatings can be formed using solvent-based and hot-melt coating processes.
- solvent-based processes the coating is made by first forming a solution or suspension comprising the solvent, the coating material and optional coating additives.
- the coating materials may be completely dissolved in the coating solvent, or only dispersed in the solvent as an emulsion or suspension or a combination of the two.
- Latex dispersions are an example of an emulsion or suspension that may be useful as in a solvent-based coating process.
- the solvent is a liquid at room temperature.
- Coating may be conducted by conventional techniques, such as by pan coaters, rotary granulators and fluidized bed coaters such as top-spray, tangential- spray or bottom-spray (Wurster coating).
- a top-spray method can also be used to apply the coating.
- coating solution is sprayed down onto the fluidized cores.
- the solvent evaporates from the coated cores and the coated cores are re- fluidized in the apparatus. Coating continues until the desired coating thickness is achieved.
- Compositions and methods for making the multiparticulates of this embodiment are detailed in the following US Patent Applications, US 2005-0181062, US 2005-0181062, US 2008-0199527, US 2005-0186285A1 which are herein incorporated as reference in their entirety.
- the multiparticulates of the invention generally are of a mean diameter from about 40 to about 3,000 micron, with a preferred range of 50 to 1 ,000 micron, and most preferably from about 100 to 300 micron. While the multiparticulates can have any shape and texture, it is preferred that they be spherical, with a smooth surface texture. These physical characteristics of the multiparticulates improve their flow properties, permit them to be uniformly coated (if desired). As used herein, the term "about” means +/- 10% of the value.
- the multiparticulates of the present invention are particularly suitable for controlled release or delayed release or any combination of these two release profiles when introduced to a use environment.
- a "use environment" can be either the in vivo environment of the gastrointestinal (Gl) tract or the in vitro dissolution tests described herein.
- Information about in vivo release rates can be determined from the pharmacokinetic profile using standard deconvolution or Wagner-Nelson treatment of the data which should be readily known to those skilled in the art.
- the latrepirdine matrix multiparticulates may be blended with compressible excipients such as lactose, microcrystalline cellulose, dicalcium phosphate, and the like and the blend compressed to form a tablet.
- compressible excipients such as lactose, microcrystalline cellulose, dicalcium phosphate, and the like
- Disintegrants such as sodium starch glycolate or crosslinked polyvinyl pyrrolidone
- Tablets prepared by this method disintegrate when placed in an aqueous medium (such as the Gl tract), thereby exposing the multiparticulate matrix which releases latrepirdine there from.
- excipients may be employed in the controlled release portion of the invention, including those excipients well known in the art, e.g., as described in Remington: The Science and Practice of Pharmacy, 20 th edition (2000).
- excipients such as surfactants, pH modifiers, fillers, matrix materials, complexing agents, solubilizers, pigments, lubricants, glidants, flavorants, and so forth may be used for customary purposes and in typical amounts without adversely affecting the properties of the compositions.
- Example matrix materials, fillers, or diluents include lactose, mannitol, xylitol, dextrose, sucrose, sorbitol, compressible sugar, microcrystalline cellulose, powdered cellulose, starch, pregelatinized starch, dextrates, dextran, dextrin, dextrose, maltodextrin, calcium carbonate, dibasic calcium phosphate, tribasic calcium phosphate, calcium sulfate, magnesium carbonate, magnesium oxide, poloxamers, polyethylene oxide, hydroxypropyl methyl cellulose and mixtures thereof.
- the controlled release portion comprises:
- the total amount of latrepirdine in the dosage form may range from 1 mg to 100 mg, preferably 15 mg to 85 mg.
- latrepirdine is incorporated into an osmotic delivery devices or "osmotic pumps" as they are known in the art.
- Osmotic pumps comprise a core containing an osmotically effective composition surrounded by a semipermeable membrane.
- semipermeable in this context means that water can readily diffuse through the membrane, but solutes dissolved in water typically cannot.
- the device imbibes water due to the osmotic activity of the core composition.
- the contents of the device cannot pass through the non-porous regions of the membrane and are driven by osmotic pressure to leave the device through an opening or passageway pre-manufactured into the dosage form or, alternatively, formed in situ in the Gl tract as by the bursting of intentionally-incorporated weak points in the coating under the influence of osmotic pressure.
- the osmotically effective composition includes water- soluble species, which generate a colloidal osmotic pressure, and water-swellable polymers. Examples of such dosage forms are well known in the art. See, for example, Remington: The Science and Practice of Pharmacy, 21 st Edition, 2006 Chapter 47; page 950-1 and herein incorporated as reference.
- latrepirdine is incorporated into a bilayer osmotic delivery device such that the latrepirdine-containing composition must include an entraining agent in the form of a water-swellable polymer and a second push layer or water swelling layer which contains water-swellable polymers and/or osmoticallly active agents, but does not contain any active agent.
- the bilayer tablet is surrounded by a semi-permeable membrane which contains one or more openings which are manufactured into the dosage form through such techniques as laser drilling.
- Such water-swellable polymers are often referred to in the pharmaceutical arts as an "osmopolymer” or a “hydrogel.”
- the entraining agent suspends or entrains the drug so as to aid in the delivery of the drug through the delivery port(s). While not wishing to be bound by any particular theory, it is believed that upon the imbibition of water into the dosage form, the entraining agent has enough viscosity to allow it to suspend or entrain the drug, while at the same time remaining sufficiently fluid to allow the entraining agent to pass through the delivery port(s) along with the drug.
- the amount of the entraining agent present in the latrepirdine-containing composition may range from about 20 wt % to about 80 wt %.
- the entraining agent may be a single material or a mixture of materials.
- Non-crosslinked polyethylene oxide (PEO) may be used as the entraining agent.
- Other suitable entraining agents include hydroxypropyl cellulose (HPC), hydroxypropylmethyl cellulose (HPMC), methylcellulose (MC), hydroxyethyl cellulose (HEC) and polyvinyl pyrrolidone (PVP), as well as mixtures of these polymers with PEO.
- the choice of the molecular weight for the PEO depends in part on whether the PEO makes up the bulk of the non-latrepirdine portion of the latrepirdine- containing composition, or whether significant amounts of other low-molecular weight water-soluble excipients are included; that is, the PEO molecular weight choice depends on the fraction of the latrepirdine-containing composition that is PEO. Should the latrepirdine-containing composition not become fluid rapidly, the dosage form can swell and rupture the coating that surrounds the core, potentially causing failure of the dosage form.
- the excipients of the latrepirdine-containing composition are primarily PEO (e.g., PEO makes up about 60 wt % or more of the non-latrepirdine components of the latrepirdine-containing composition), it is generally preferred that the PEO have an average molecular weight of from about 100,000 to 300,000 daltons. (As used herein, reference to molecular weights of polymers should be taken to mean average molecular weights.)
- another embodiment of the present invention uses a higher molecular weight of PEO from about 500,000 to 800,000 daltons at a lower fraction of the non-latrepirdine excipients, a portion of the PEO being replaced with a fluidizing agent.
- PEO makes up about 60 wt % or more of the non-latrepirdine components of the latrepirdine-containing composition
- PEO having a molecular weight of 500,000 daltons or more makes the latrepirdine-containing composition too viscous, and can result in a rupture of the coating or at least in a delay of the release of latrepirdine.
- the non-latrepirdine components of the latrepirdine-containing composition comprise less than about 60 wt % PEO and also contain a fluidizing agent.
- the amount of fluidizing agent present in the latrepirdine-containing composition may range from about 5 to about 50 wt %, preferably 10 to 30 wt % of the latrepirdine-containing composition.
- Preferred fluidizing agents are low molecular weight, water-soluble solutes such as non-reducing sugars and organic acids with aqueous solubilities of 30 img/mL or greater.
- Suitable sugars include xylitol, mannitol, sorbitol, and maltitol.
- Salts useful as a fluidizing agent include sodium chloride, sodium lactate and sodium acetate.
- Organic acids useful as a fluidizing agent include adipic acid, citric acid, malic acid, fumaric acid, succinic acid and tartaric acid.
- the latrepirdine-containing composition may also contain other water- swellable polymers.
- the latrepirdine-containing composition may contain relatively small amounts of water-swellable polymers that greatly expand in the presence of water.
- water-swellable polymers include sodium starch glycolate, sold under the trade name EXPLOTAB, and croscarmelose sodium, sold under the trade name AC-DI-SOL.
- Such polymers may be present in amounts ranging from 0 wt % to 10 wt % of the latrepirdine-containing composition.
- the latrepirdine-containing composition may optionally include osmotically effective solutes, often referred to as "osmogens" or "osmagents.”
- the amount of osmagent present in the latrepirdine-containing composition may range from about 0 wt % to about 50 wt %, preferably 10 wt % to 30 wt % of the latrepirdine-containing composition.
- suitable osmagents are water-soluble salts, sugars, organic acids, and other low-molecule-weight organic compounds that are capable of imbibing water to thereby establish an osmotic pressure gradient across the barrier of the surrounding coating.
- Typical useful salts include magnesium sulfate, magnesium chloride, calcium chloride, sodium chloride, lithium chloride, potassium sulfate, sodium carbonate, sodium sulfite, lithium sulfate, potassium chloride, and sodium sulfate.
- chloride salts such as sodium chloride are utilized as osmagents.
- the latrepirdine-containing composition may further include solubility- enhancing agents or solubilizers that promote the aqueous solubility of the drug, present in an amount ranging from about 0 to about 30 wt % of the latrepirdine- containing composition.
- Solubilizers useful with latrepirdine include organic acids and organic acid salts, partial glycerides, e.g., less than fully esterified derivatives of glycerin, including glycerides, monoglycerides, diglycerides, glyceride derivatives, polyethylene glycol esters, polypropylene glycol esters, polyhydric alcohol esters, polyoxyethylene ethers, sorbitan esters, polyoxyethylene sorbitan esters, and carbonate salts.
- partial glycerides e.g., less than fully esterified derivatives of glycerin, including glycerides, monoglycerides, diglycerides, glyceride derivatives, polyethylene glycol esters, polypropylene glycol esters, polyhydric alcohol esters, polyoxyethylene ethers, sorbitan esters, polyoxyethylene sorbitan esters, and carbonate salts.
- a preferred class of solubilizers is organic acids. Since latrepirdine is a base which is solubilized by protonation, and since its solubility in an aqueous environment of pH 5 or higher is reduced, it is believed that addition of an organic acid to the Latrepirdine-containing composition assists in solubilization and hence absorption of latrepirdine. Even a slight decrease in the pH of the aqueous solution at high pH results in dramatic increases in the solubility of latrepirdine. Organic acids can also promote stability during storage prior to introduction to a use environment due to their tendency to maintain latrepirdine in a protonated state.
- organic acids meeting such criteria consists of citric, succinic, fumaric, adipic, malic and tartaric acids.
- Citric, malic, and tartaric acid have the advantage of high water solubility and high osmotic pressure.
- Succinic and fumaric acid offer a combination of both moderate solubility and moderate osmotic pressure.
- the water-swellable composition may also optionally contain a colorant.
- the purpose of the colorant is to allow identification of the drug-containing side of the tablet face for purposes of providing the delivery port, such as by laser drilling through the coating.
- Acceptable colorants include, but are not limited to, Red Lake No. 40, FD C Blue 2 and FD C Yellow 6.
- Both the Latrepirdine-containing composition and/or the water-swellable composition may optionally contain an antioxidant, such as but not limited to BHT, vitamin E, BHA, or ascorbyl palmitate.
- the antioxidant may be present in an amount ranging from 0 to 1 wt % of the Latrepirdine-containing and/or the water-swellable composition.
- Water-swellable composition may also include other conventional pharmaceutically useful excipients such as a binder, including HPC, HPMC, HEC, MC, and PVP, a tableting aid, such as microcrystalline cellulose, and a lubricant such as magnesium stearate.
- a binder including HPC, HPMC, HEC, MC, and PVP
- a tableting aid such as microcrystalline cellulose
- a lubricant such as magnesium stearate.
- the water-swellable composition is prepared by mixing the water-swellable polymer and the other excipients to form a uniform blend. To obtain a uniform blend, it is desirable to either wet or dry granulate or dry blend ingredients that have similar particle sizes using the types of processes known to those skilled in the art.
- the core is prepared by first placing a mixture of the latrepirdine-containing composition into a tablet press and then leveling the mixture by gentle compression.
- the water-swellable composition is then placed on top of the latrepirdine-containing composition and compressed in order to complete formation of the core.
- the water-swellable composition can be placed into the tablet press first, followed by the latrepirdine-containing composition.
- the respective amounts of latrepirdine-containing composition and water- swellable composition are chosen to provide satisfactory latrepirdine release.
- it is desired to provide a large latrepirdine dose in a relatively small dosage size it is desired to maximize the amount of latrepirdine-containing composition and minimize the amount of water-swellable composition, while still obtaining good release performance.
- the latrepirdine-containing composition may comprise from about 50 to about 85 wt % of the core, and preferably from about 60 to about 70 wt %.
- the latrepirdine-containing composition may comprise from 50 to 90 wt % of the core, and preferably from about 75 to about 85 wt %. Those values correspond to the weight ratio of the latrepirdine-containing composition to water-swellable composition of from 1 to 9.
- the absolute value of the diameter and height of the tablets of the present invention can vary over a wide range.
- the semi-permeable coating is applied.
- the coating should have high water permeability and a high strength, while at the same time be easily fabricated and applied. High water permeability is required to permit water to enter the core in sufficient volume. High strength is required to ensure the coating does not burst when the core swells as it imbibes water, leading to an uncontrolled delivery of the core contents. Finally, the coating must have high reproducibility and yield.
- the coating have at least one delivery port in communication with the interior and exterior of the coating for delivery of the latrepirdine-containing composition.
- the coating must be non-dissolving and non-eroding during release of the latrepirdine-containing composition, generally meaning that it be water-insoluble, such that latrepirdine is substantially entirely delivered through the delivery port(s), in contrast to delivery via permeation through the coating.
- Coatings with these characteristics can be obtained using hydrophilic polymers such as plasticized and unplasticized cellulose esters, ethers, and ester- ethers.
- Particularly suitable polymers include cellulose acetate (CA), cellulose acetate butyrate (CAB), and ethyl cellulose (EC).
- CA cellulose acetate
- CAB cellulose acetate butyrate
- EC ethyl cellulose
- One set of polymers are cellulose acetates having acetyl contents of 25 to 42%.
- One typical polymer is CA having an acetyl content of 39.8%, specifically, CA 398-10 (Eastman Fine Chemicals, Kingsport, Tenn.). CA 398-10 is reported to have an average molecular weight of about 40,000 daltons.
- CA having an acetyl content of 39.8% is high molecular weight CA having an average molecular weight greater than about 45,000, and specifically, CA 398-30 (Eastman Fine Chemical) which is reported to have an average molecular weight of 50,000 daltons.
- Coating is conducted in conventional fashion by first forming a coating solution and then coating by dipping, fluidized bed coating, or by pan coating. To accomplish this, a coating solution is formed comprising the polymer and a solvent.
- Typical solvents useful with the cellulosic polymers above include acetone, methyl acetate, ethyl acetate, isopropyl acetate, n-butyl acetate, methyl isobutyl ketone, methyl propyl ketone, ethylene glycol monoethyl ether, ethylene glycol monoethyl acetate, methylene dichloride, ethylene dichloride, propylene dichloride, nitroethane, nitropropane, tetrachloroethane, 1 ,4-dioxane, tetrahydrofuran, diglyme, and mixtures thereof.
- the coating solution typically contains 3 to 15 wt % of the polymer.
- the coating solution may also include pore-formers or non-solvents in any amount as long as the polymer remains soluble at the conditions used to form the coating and as long as the coating remains water permeable and has sufficient strength. Pore-formers and their use in fabricating coatings are described in U.S. Pat. Nos. 5,698,220 and 5,612,059, the pertinent disclosures of which are incorporated herein by reference.
- pore former refers to a material added to the coating solution that has low or no volatility relative to the solvent such that it remains as part of the coating following the coating process but that is sufficiently water swellable or water soluble such that, in the aqueous use environment it provides a water-filled or water-swollen channel or "pore” to allow the passage of water, thereby enhancing the water permeability of the coating.
- Suitable pore formers include but are not limited to hydroxypropylcellulose (HPC), polyethylene glycol (“PEG”), PVP, and PEO.
- the weight ratio of CA:PEG or CA:HPC should range from about 6:4 to about 9:1 .
- a non-solvent such as water
- non-solvent any material added to the coating solution that substantially dissolves in the coating solution and reduces the solubility of the coating polymer or polymers in the solvent.
- the function of the non-solvent is to impart porosity to the resulting coating.
- porous coatings have higher water permeability than an equivalent weight of a coating of the same composition that is not porous and this porosity is indicated by a reduction in the density of the coating (mass/volume).
- a non-solvent imparts porosity to the coating during evaporation of solvent by causing the coating solution to undergo liquid and liquid phase separation prior to solidification.
- the suitability and amount of a particular candidate material can be evaluated for use as a non-solvent by progressively adding the candidate non- solvent to the coating solution until it becomes cloudy.
- the cloud point an appropriate level of non-solvent for maximum porosity is the amount just below the cloud point.
- the cloud point is at about 23 wt % water.
- Suitable non-solvents are any materials that have appreciable solubility in the solvent and that lower the coating polymer solubility in the solvent.
- the preferred non-solvent depends on the solvent and the coating polymer chosen.
- suitable non-solvents include water, glycerol, alcohols such as methanol or ethanol.
- coating solution weight ratios of CA:PEG 3350: water are 7:3:5, 8:2:5, and 9:1 :5, with the remainder of the solution comprising a solvent such as acetone.
- a solvent such as acetone.
- Coatings formed from these coating solutions are generally porous.
- porous is meant that the coating in the dry state has a density less than the density of the same material in a nonporous form.
- nonporous form is meant a coating material formed by using a coating solution containing no non-solvent, or the minimal amount of non-solvent required to produce a homogeneous coating solution.
- the dry-state density of the coating can be calculated by dividing the coating weight (determined from the weight gain of the tablets before and after coating) by the coating volume (calculated by multiplying the coating thickness, as determined by optical or scanning electron microscopy, by the tablet surface area).
- the porosity of the coating is one of the factors that leads to the combination of high water permeability and high strength of the coating.
- the weight of the coating around the core depends on the composition and porosity of the coating, but generally should be present in an amount ranging from 3 to 30 wt %, based on the weight of the uncoated core.
- porous coatings based on CA, PEG, and water described above translate to excellent results
- other pharmaceutically acceptable materials could be used in the coating so long as the coating has the requisite combination of high water permeability, high strength, and ease of fabrication and application.
- coatings may be dense, porous, or "asymmetric,” having one or more dense layers and one or more porous layers such as those disclosed in U.S. Pat. Nos. 5,612,059 and 5,698,220, the pertinent disclosures of which are incorporated herein by reference.
- the coating must also contain at least one delivery port in communication with the interior and exterior of the coating to allow for release of the drug-containing composition to the exterior of the dosage form.
- the delivery port can range in size from about the size of the drug particles, and thus could be as small as 1 to 100 microns in diameter and may be termed pores, up to about 5000 microns in diameter.
- the shape of the port may be substantially circular, in the form of a slit, or other convenient shape to ease manufacturing and processing.
- the port(s) may be formed by post-coating mechanical or thermal means or with a beam of light (e.g., a laser), a beam of particles, or other high-energy source, or may be formed in situ by rupture of a small portion of the coating.
- Delivery ports may also be formed in situ by erosion of a plug of water-soluble material or by rupture of a thinner portion of the coating over an indentation in the core. Delivery ports may be formed by coating the core such that one or more small regions remains uncoated.
- the delivery port can be a large number of holes or pores that may be formed during coating, as in the case of asymmetric membrane coatings, described in more detail herein, and of the type disclosed in U.S. Pat. Nos. 5,612,059 and 5,698,220, the disclosures of which are incorporated by reference. When the delivery pathways are pores there can be a multitude of such pores that range in size from 1 micron to greater than 100 microns.
- At least one delivery port should be formed on the side of coating that is adjacent to the latrepirdine-containing composition, so that the latrepirdine-containing composition will be extruded out of the delivery port by the swelling action of the water-swellable composition. It is recognized that some processes for forming delivery ports may also form holes or pores in the coating adjacent to the water- swellable composition.
- the coating may optionally include a port in communication with the water- swellable composition.
- a delivery port does not typically alter the latrepirdine release characteristics of the dosage form, but may provide manufacturing advantages. It is believed that the water-swellable compositions, such as those containing PEO with a molecular weight between 3,000,000 and 8,000,000 daltons, are too viscous to appreciably exit the port.
- the tablet In dosage forms wherein the delivery ports are drilled either mechanically or by laser, the tablet must be oriented so that at least one delivery port is formed in the coating adjacent to the latrepirdine-containing composition.
- a colorant within the water-swellable composition is used to orient the core dosage form during the drilling step in manufacture. By providing a delivery port on both faces of the dosage form, the need to orient the dosage form may be eliminated and the colorant may be removed from the water-swellable composition.
- latrepirdine is incorporated into a variation of the above disclosed osmotic delivery device, an asymmetric membrane technology (AMT).
- AMT asymmetric membrane technology
- porous coatings In the formation of these porous coatings, a water-insoluble polymer is combined with a water- soluble, pore-forming material. The mixture is coated onto an osmotic tablet core from a combination of water and solvent. As the coating dries, a phase inversion process occurs whereby a porous, asymmetric membrane is produced.
- AMT system for controlled release of a drug with similar physiochemical properties is described in US Patent Application Publication US2007/0248671 and herein incorporated as reference.
- One aspect of the present invention provides a dosage form which comprises (a) a core containing at least one pharmaceutically active ingredient and (b) at least one asymmetric membrane technology coating wherein said coating comprises: a. one or more substantially water-insoluble polymers, and
- One aspect of the present invention also provides a dosage form wherein the dosage form delivers drug primarily by osmotic pressure.
- the present invention provides a dosage form wherein the pharmaceutically active ingredient is latrepirdine or a pharmaceutically acceptable salt thereof.
- the water- insoluble polymer as used in the present invention preferably comprises a cellulose derivative, more preferably, cellulose acetate.
- the solid, water-soluble polymeric material as used in the present invention comprises a polymer having a weight average molecular weight between 2000 and 50,000 daltons.
- the solid, water-soluble polymeric material is selected from the group consisting of water-soluble cellulose derivatives, acacia, dextrin, guar gum, maltodextrin, sodium alginate, starch, polyacrylates, polyvinyl alcohols and zein.
- the water-soluble cellulose derivatives comprise hydroxypropylcellulose, hydroxypropylmethylcellulose and hydroxyethylcellulose.
- the solid, water-soluble, polymeric material has a viscosity for a 5 percent w:w aqueous solution of less than 400 mPa s.
- the solid, water-soluble, polymeric material has a viscosity for a 5 percent w:w aqueous solution of less than 300 mPa s. In other embodiments, the solid, water-soluble, polymeric material has a softening temperature greater than 55 degrees C.
- the dosage form of the present invention may be a tablet or a multiparticulate.
- the core of the present invention contains a sugar. More preferably, the sugar is mannitol.
- the water- insoluble polymer is cellulose acetate and said solid, water-soluble polymeric material is hydroxypropylcellulose.
- the dosage form of the invention contains latrepirdine, or a pharmaceutically acceptable salt thereof, as the pharmaceutically active ingredient, while the water-insoluble polymer is cellulose acetate and the solid, water-soluble polymeric material is hydroxypropylcellulose.
- a process of the present invention encompasses the process wherein the coating is applied from a mixture of acetone and water using a pan coating.
- the process of the present invention also encompasses the process wherein the asymmetric membrane comprises cellulose acetate and hydroxypropylcellulose which is coated from a mixture of acetone to water between about 9:1 and 6:4, w:w, and more preferably between about 3: 1 and about 4.5: 1 , w:w, using a pan coater.
- the process of the present invention encompasses the process wherein the core comprises latrepirdine, or a pharmaceutically acceptable salt thereof.
- the water-insoluble component of the asymmetric membrane coating preferentially is formed from cellulose derivatives.
- these derivatives include cellulose esters and ethers, namely the mono-, di- and triacyl esters wherein the acyl group consists of two to four carbon atoms and lower alkyl ethers of cellulose wherein the alkyl group has one to four carbon atoms.
- the cellulose esters can also be mixed esters, such as cellulose acetate butyrate, or a blend of cellulose esters. The same variations can be found in ethers of cellulose and include blends of cellulose esters and cellulose ethers.
- cellulose derivatives which can be used in making asymmetric membranes of the present invention include cellulose nitrate, acetaldehyde dimethyl cellulose, cellulose acetate ethyl carbamate, cellulose acetate phthalate, cellulose acetate methyl carbamate, cellulose acetate succinate, cellulose acetate dimethaminoacetate, cellulose acetate ethyl carbonate, cellulose acetate dimethaminoacetate, cellulose acetate ethyl carbonate, cellulose acetate chloroacetate, cellulose acetate ethyl oxalate, cellulose acetate methyl sulfonate, cellulose acetate butyl sulfonate, cellulose acetate p-toluene sulfonate, cellulose cyanoacetates, cellulose acetate trimellitate, cellulose methacrylates and hydroxypropylmethylcellulose acetate succinate.
- a particularly preferred water- insoluble component is cellulose acetate.
- Particularly preferred cellulose acetates include those having an acetyl content of about 40 percent and a hydroxyl content of about 3.5 percent.
- Other materials also can be used in the fabrication of asymmetric membrane technology coatings, provided such materials are substantially water- insoluble, film-forming and safe to use in pharmaceutical applications.
- yhe water-soluble polymeric component of the present invention comprises solid, polymeric materials that do not form hydrogen peroxide or formaldehyde upon storage for 12 weeks at 40 degrees C./75 percent relative humidity, in an amount greater than about 0.01 percent w/w (100 parts per million, ppm).
- the solid polymeric water-soluble material preferentially has a water- solubility of greater than 0.5 img/mL; more preferably, greater than 2 img/mL; and still more preferably, greater than 5 img/mL
- the solid polymeric water-soluble material has a melting or softening temperature above room temperature.
- the solid material has a melting or softening temperature above 30 degrees C; more preferentially, above 40 degrees C; and most preferentially, above 50 degrees C.
- Melting and softening points can be determined visually using a melting point apparatus, or alternatively, can be measured using differential scanning calorimetry (DSC), as is known in the art.
- the polymer can be either a homopolymer or a copolymer. Such polymers can be natural polymers, or be derivatives of natural products, or be entirely synthetic. The molecular weight of such materials is preferentially high enough to prevent migration and aid in film-forming, yet low enough to allow coating (as discussed below).
- the preferred molecular weight range for the present invention is therefore between 2000 and 50,000 daltons (weight average).
- Preferred polymers suitable as water-soluble components of an asymmetric membrane technology coating for the present invention include substituted, water-soluble cellulose derivatives, acacia, dextrin, guar gum, maltodextrin, sodium alginate, starch, polyacrylates, polyvinyl alcohols and zein.
- Particularly preferred water-soluble polymers include hydroxyethylcellulose, hydroxypropylcellulose and polyvinylalcohol.
- the viscosity of the coating solution is too high, and that one approach to solving this issue is to use more dilute solutions of the polymer. Due to the phase behavior of the coating solution, having both water-soluble and organic-soluble components, there is a limit to how low the concentration of the water-soluble polymer can be and still provide a commercializable process. For this reason, it is preferred that the water-soluble polymers not have too high a viscosity. Viscosities can be determined at 25 degrees C. using a Brookfield LVF viscometer (available from Brookfield Engineering Corp., Middleboro, Mass.) with spindle and speed combinations depending on viscosity levels for 5 percent (w:w) aqueous solutions. Preferred water-soluble polymers have viscosities for 5 percent (w:w) solutions of less than 400 mPa s; more preferably, less than 300 mPa s.
- especially preferred water-soluble polymers include hydroxypropylcellulose and hydroxyethylcellulose having a viscosity for a 5 percent (w:w) of less than 300 mPa s.
- Commercially available examples of such polymers include Klucel EF.TM. and Natrasol LR.TM., both made by the Aqualon Division of Hercules Corp., Hopewell, Va.
- the water-soluble, solid polymeric material's stability to formation of hydrogen peroxide can be measured by storing the polymer in an oven having a temperature and relative humidity (RH) of 40 degrees C. and 75 percent RH, respectively.
- the polymer should be stored exposed to the oven environment under "open” conditions.
- the polymer should be stored for at least 12 weeks.
- Levels of hydrogen peroxide can be administered as described in G. M. Eisenberg, "Colorimetric determination of hydrogen peroxide” in Ind. Eng. Chem. (Anal. Ed.), 1943, 15, 327-328. Under these storage conditions, acceptable polymeric materials for the present invention have hydrogen peroxide levels below 100 parts per million (ppm); more preferably, below 50 ppm; and most preferably, below 10 ppm.
- the water-soluble polymer's stability to formation of formaldehyde can be measured by storing the polymer in an oven at 40 degrees C. and 75 percent RH. Polymer should be stored in a sealed container to avoid loss of volatile formaldehyde. The polymer should be stored for at least 12 weeks. Levels of formaldehyde can be determined as described in M. Ashraf-Khorassani, et al., "Purification of pharmaceutical excipients with supercritical fluid extraction” in Pharm. Dev. Tech. 2005, 10, 1-10. Under these storage conditions, acceptable water-soluble polymeric materials for the present invention have formaldehyde levels below 100 ppm, more preferably, below 50 ppm, and most preferably, below 10 ppm.
- the asymmetric membrane technology coating formulation can contain small amounts of other materials without significantly changing its function or altering the nature of the present invention.
- additives include glidants (e.g., talc and silica) and plasticizers (e.g., triethylcitrate and triacetin), which are typically added, when needed, at levels of less than about 5 percent (w:w) of the coating.
- active pharmaceutical ingredients can also be in the form of pharmaceutically acceptable salts.
- the cores for the present invention can also employ solubilizing additives.
- Such additives include pH-buffering additives to maintain the core at a pH wherein the active pharmaceutical ingredient has a sufficiently high solubility to be pumped out of the dosage form in solution.
- the active pharmaceutical ingredient can be present in the core at levels ranging from about 0.1 percent (w:w) to about 75 percent (w:w).
- the core can contain osmotic agents which help to provide the driving force for drug delivery.
- osmotic agents include water-soluble sugars and salts.
- a particularly preferred osmotic agent is mannitol or sodium chloride.
- the core of the AMT system can contain other additives to provide for such benefits as stability, manufacturability and system performance.
- Stabilizing excipients include pH-modifying ingredients, antioxidants, chelating agents, and other such additives as is known in the art.
- Excipients that improve manufacturability include agents to help in flow, compression or extrusion. Flow can be helped by such additives as talc, stearates and silica. Flow is also improved by granulation of the drug and excipients, as is known in the art. Such granulations often benefit from the addition of binders such as hydroxypropylcellulose, starch and polyvinylpyrollidone (povidone).
- binders such as hydroxypropylcellulose, starch and polyvinylpyrollidone (povidone).
- Compression can be improved by the addition of diluents to the formulation.
- diluents include lactose, mannitol, microcrystalline cellulose and the like, as is known in the art.
- the melt properties of the excipients can be important. Generally, it is preferable that such excipients have melting temperatures below about 100 degrees C.
- appropriate excipients for melt processes include esterified glycerines and stearyl alcohol.
- manufacturability can be improved by addition of lubricants.
- a particularly preferred lubricant is magnesium stearate.
- Cores can be produced using standard tablet compression processes, as is known in the art. Such processes involve powders filling dies followed by compression using appropriate punches. Cores can also be produced by an extrusion process. Extrusion processes are especially well-suited to making small cores (multiparticulates). A preferred extrusion process is a melt-spray-congeal process as described in WO2005/053653A1 , incorporated by reference. Cores can also be prepared by layering drug onto seed cores. Such seed cores are preferentially made of sugar or microcrystalline cellulose. Drug can be applied onto the cores by spraying, preferentially in a fluid-bed operation, as is known in the art.
- the cores are coated with the asymmetric membrane by any technique that can provide the asymmetric membrane as a coating over the entire cores.
- Preferred coating methods include pan coating and fluid-bed coating.
- the water-insoluble polymer and water-soluble polymer as well as any other additives are first dissolved or dispersed in an appropriate solvent or solvent combination.
- the coating solvent needs to be optimized for performance.
- the solvents are chosen such that the more volatile solvent is the better solvent for the water-insoluble polymeric component. The result is that during coating, the water-insoluble polymeric component precipitates from solution.
- Preferred solvents and solvent ratios can be determined by examining the multi- component solubility behavior of the system.
- a preferred solvent mixture is acetone and water, with a ratio of between about 9: 1 and about 6:4, w:w.
- latrepirdine sustained-release dosage forms of this invention includes membrane-moderated or reservoir systems.
- a reservoir of latrepirdine is surrounded by a rate-limiting membrane.
- the latrepirdine traverses the membrane by mass transport mechanisms well known in the art, including but not limited to dissolution in the membrane followed by diffusion across the membrane or diffusion through liquid-filled pores within the membrane.
- These individual reservoir system dosage forms may be large, as in the case of a tablet containing a single large reservoir, or multiparticulate, as in the case of a capsule containing a plurality of reservoir particles, each individually coated with a membrane.
- the coating can be non-porous, yet permeable to latrepirdine (for example latrepirdine may diffuse directly through the membrane), or it may be porous.
- the particular mechanism of transport is not believed to be critical.
- Sustained release coatings as known in the art may be employed to fabricate the membrane, especially polymer coatings, such as a cellulose ester or ether, an acrylic polymer, or a mixture of polymers.
- Preferred materials include ethyl cellulose, cellulose acetate and cellulose acetate butyrate.
- the polymer may be applied as a solution in an organic solvent or as an aqueous dispersion or latex.
- the coating operation may be conducted in standard equipment such as a fluid bed coater, a Wurster coater, or a rotary bed coater.
- the permeability of the coating may be adjusted by blending of two or more materials.
- a useful process for tailoring the porosity of the coating comprises adding a pre-determined amount of a finely-divided water-soluble material, such as sugars or salts or water-soluble polymers to a solution or dispersion (e.g., an aqueous latex) of the membrane-forming polymer to be used.
- a solution or dispersion e.g., an aqueous latex
- the membrane coating can also be modified by the addition of plasticizers, as known in the art.
- a useful variation of the process for applying a membrane coating comprises dissolving the coating polymer in a mixture of solvents chosen such that as the coating dries, a phase inversion takes place in the applied coating solution, resulting in a membrane with a porous structure.
- a phase inversion takes place in the applied coating solution, resulting in a membrane with a porous structure.
- the morphology of the membrane is not of critical importance so long as the permeability characteristics enumerated herein are met.
- the membrane can be amorphous or crystalline. It can have any category of morphology produced by any particular process and can be, for example, an interfacially-polymerized membrane (which comprises a thin rate-limiting skin on a porous support), a porous hydrophilic membrane, a porous hydrophobic membrane, a hydrogel membrane, an ionic membrane, and other such materials which are characterized by controlled permeability to latrepirdine.
- a useful reservoir system embodiment is a capsule having a shell comprising the material of the rate-limiting membrane, including any of the membrane materials previously discussed, and filled with a latrepirdine drug composition.
- a particular advantage of this configuration is that the capsule may be prepared independently of the drug composition, thus process conditions that would adversely affect the drug can be used to prepare the capsule.
- One embodiment is a capsule having a shell made of a porous or a permeable polymer made by a thermal forming process.
- Another embodiment is a capsule shell in the form of an asymmetric membrane; e.g., a membrane that has a thin skin on one surface and most of whose thickness is constituted of a highly permeable porous material.
- a process for preparation of asymmetric membrane capsules comprises a solvent exchange phase inversion, wherein a solution of polymer, coated on a capsule-shaped mold, is induced to phase-separate by exchanging the solvent with a-miscible non-solvent.
- asymmetric membranes useful in this invention are disclosed in the aforementioned European Patent Specification 0 357 369 B1.
- Another embodiment of the class of reservoir systems comprises a multiparticulate wherein each particle is coated with a polymer designed to yield sustained release of latrepirdine.
- the multiparticulate particles each comprise latrepirdine and one or more excipients as needed for fabrication and performance.
- the size of individual particles is generally between about 50 micron and about 3 mm, although beads of a size outside this range may also be useful.
- the beads comprise latrepirdine and one or more binders. As it is generally desirable to produce dosage forms which are small and easy to swallow, beads which contain a high fraction of latrepirdine relative to excipients are preferred.
- Binders useful in fabrication of these beads include microcrystalline cellulose (e.g., Avicel.RTM., FMC Corp.), hydroxypropyl cellulose (HPC), hydroxypropyl methyl cellulose (HPMC), and related materials or combinations thereof.
- binders which are useful in granulation and tabletting such as starch, pregelatinized starch, and poly (N-vinyl-2-pyrrolidinone) (PVP) may also be used to form multiparticulates.
- Reservoir system latrepirdine multiparticulates may be prepared using techniques known to those skilled in the art, including, but not limited to, the techniques of extrusion and spheronization, wet granulation, fluid bed granulation, and rotary bed granulation.
- the beads may also be prepared by building the latrepirdine composition (drug plus excipients) up on a seed core (such as a nonpareil seed) by a drug-layering technique such as powder coating or by applying the latrepirdine composition by spraying a solution or dispersion of latrepirdine in an appropriate binder solution onto seed cores in a fluidized bed such as a Wurster coater or a rotary processor.
- a suitable composition and method is to spray a dispersion of a latrepirdine/hydroxypropylcellulose composition in water.
- latrepirdine can be loaded in the aqueous composition beyond its solubility limit in water.
- a method for manufacturing the multiparticulate cores of this embodiment is the extrusion/spheronization process, as previously discussed for matrix multiparticulates.
- Another process and composition for this method involves using water to wet-mass blend of about 5 to 75% of micro-crystalline cellulose with correspondingly about 95 to 25% latrepirdine.
- the process involves the use of water to wet-mass blend of about 5-30% microcrystalline cellulose with correspondingly about 5-70% latrepirdine.
- a sustained release coating as known in the art, especially polymer coatings, may be employed to fabricate the membrane, as previously discussed for reservoir systems. Suitable and preferred polymer coating materials, equipment, and coating methods also include those previously discussed.
- the rate of latrepirdine release from the coated multiparticulates can also be controlled by factors such as the composition and binder content of the drug- containing core, the thickness and permeability of the coating, and the surface-to- volume ratio of the multiparticulates. It will be appreciated by those skilled in the art that increasing the thickness of the coating will decrease the release rate, whereas increasing the permeability of the coating or the surface-to-volume ratio of the multiparticulates will increase the release rate. If desired, the permeability of the coating may be adjusted by blending of two or more materials.
- a useful series of coatings comprises mixtures of water-insoluble and water-soluble polymers, for example, ethylcellulose and hydroxypropyl methylcellulose, respectively.
- a useful modification to the coating is the addition of finely-divided water-soluble material, such as sugars or salts. When placed in an aqueous medium, these water soluble membrane additives are leached out of the membrane, leaving pores which facilitate delivery of the drug.
- the membrane coating may also be modified by the addition of plasticizers, as is known to those skilled in the art.
- Another useful variation of the membrane coating utilizes a mixture of solvents chosen such that as the coating dries, a phase inversion takes place in the applied coating solution, resulting in a membrane with a porous structure.
- Another embodiment is a multiparticulate comprising about 5-50 % latrepirdine, the individual particles being coated with an aqueous dispersion of ethyl cellulose, which dries to form a continuous film.
- latrepirdine beads are less than about 400 micron in size and are coated with a phase inversion membrane of ethyl cellulose or cellulose acetate.
- latrepirdine beads are less than about 400 micron in size and are coated with an aqueous dispersion of ethyl cellulose, which dries to form a continuous film.
- latrepirdine beads are less than about 300 micron in size and are coated with film an aqueous dispersion of ethyl cellulose, which dries to form a continuous film.
- another class of dosage forms includes those forms which incorporate a delay before the onset of controlled release of latrepirdine.
- a tablet comprising a core containing latrepirdine coated with a first coating of a polymeric material of the type useful for controlled release of latrepirdine and a second coating of the type useful for delaying release of drugs when the dosage form is ingested.
- the first coating is applied over and surrounds the tablet.
- the second coating is applied over and surrounds the first coating.
- the tablet can be prepared by techniques well known in the art and contains a therapeutically useful amount of latrepirdine plus such excipients as are necessary to form the tablet by such techniques.
- the first coating may be a controlled release coating as known in the art, especially polymer coatings, to fabricate the membrane, as previously discussed for reservoir systems. Suitable polymer coating materials, equipment, and coating methods also include those previously discussed.
- Materials useful for preparing the second coating on the tablet include polymers known in the art as enteric coatings for delayed-release of pharmaceuticals. These most commonly are pH-sensitive materials such as cellulose acetate phthalate, cellulose acetate trimellitate, hydroxypropyl methyl cellulose phthalate, polyvinyl acetate phthalate), and acrylic copolymers such as Eudragit L- 100 (RohmPharma), Eudragit L 30 D-55, Eudragit S 100, Eudragit FS 30D, and related materials, as more fully detailed below under "Delayed Release". The thickness and type of the delayed-release coating is adjusted to give the desired delay property.
- thicker coatings are more resistant to erosion and, consequently, yield a longer delay as do coatings which are designed to dissolve above pH 7.
- Preferred coatings typically range from about 10 micron in thickness to about 3 mm in thickness and more preferably 10 um to 500 um.
- the twice-coated tablet passes through the stomach, where the second coating prevents release of the latrepirdine under the acidic conditions prevalent there.
- the second coating erodes or dissolves according to the physicochemical properties of the chosen material.
- the first coating prevents immediate or rapid release of the latrepirdine and modulates the release so as to prevent the production of high concentrations, thereby minimizing side-effects.
- Another embodiment comprises a multiparticulate wherein each particle is dual coated as described above for tablets, first with a polymer designed to yield controlled release of the latrepirdine and then coated with a polymer designed to delay onset of release in the environment of the Gl tract when the dosage form is ingested.
- the beads contain latrepirdine and may contain one or more excipients as needed for fabrication and performance. Multiparticulates which contain a high fraction of latrepirdine relative to binder are desired.
- the multiparticulate may be of a composition and be fabricated by any of the techniques previously disclosed for multiparticulates used to make reservoir systems (including extrusion and spheronization, wet granulation, fluid bed granulation, and rotary bed granulation, seed building, and so forth).
- the controlled release coating may be as known in the art, especially polymer coatings, to fabricate the membrane, as previously discussed for reservoir systems. Suitable polymer coating materials, equipment, and coating methods also include those previously discussed.
- the rate of latrepirdine release from the controlled-release-coated multiparticulates e.g., the multiparticulates before they receive the delayed-release coating
- methods of modifying the coating are also controlled by the factors previously discussed for reservoir system latrepirdine multiparticulates.
- the second membrane or coating for dual coated multiparticulates is a delayed-release coating which is applied over the first controlled-release coating, as disclosed above for tablets, and may be formed from the same materials.
- enteric the use of the so-called "enteric" materials to practice this embodiment differs significantly from their use to produce conventional enteric dosage forms. With conventional enteric forms, the object is to delay release of the drug until the dosage form has passed the stomach and then to deliver the dose shortly after emptying from the stomach. Dosing of latrepirdine directly and completely to the duodenum is undesirable, however, due to local metabolism which is sought to be minimized or avoided by this invention.
- sustained release osmotic systems as defined above, could also be defined in the current delay then controlled release category.
- Typical osmotic sustained release systems have an initial delay of 0.5-6 hours prior to drug release in a controlled fashion.
- a standard osmotic monolithic or bilayer sustained release system embodies the definition of delay followed by controlled release.
- a first delayed release embodiment according to the invention is a "pH- dependent coated tablet", which comprises a tablet core comprising latrepirdine, a disintegrant, a lubricant, and one or more pharmaceutical carriers, such core being coated with a material, preferably a polymer, which is substantially insoluble and impermeable at the pH of the stomach, and which is more soluble and permeable at the pH of the small intestine.
- the coating polymer is substantially insoluble and impermeable at pH 5.0.
- the tablet core be coated with an amount of polymer sufficient to assure that substantially no release of latrepirdine from the dosage form occurs until the dosage form has exited the stomach and has resided in the small intestine for about 15 minutes or greater, preferably about 30 minutes or greater, thus assuring that minimal latrepirdine is released in the duodenum.
- Mixtures of a pH-sensitive polymer with a water-insoluble polymer may also be employed. Tablets are coated with an amount of polymer at a coating coverage from about 2 to 8 mg/cm 2 .
- pH-sensitive polymers which are relatively insoluble and impermeable at the pH of the stomach, but which are more soluble and permeable at the pH of the small intestine and colon include polyacrylamides, phthalate derivatives such as acid phthalates of carbohydrates, amylose acetate phthalate, cellulose acetate phthalate, other cellulose ester phthalates, cellulose ether phthalates, hydroxypropylcellulose phthalate, hydroxypropylethylcellulose phthalate, hydroxypropylmethylcellulose phthalate, methylcellulose phthalate, polyvinyl acetate phthalate, polyvinyl acetate hydrogen phthalate, sodium cellulose acetate phthalate, starch acid phthalate, styrene-maleic acid dibutyl phthalate copolymer, styrene-maleic acid polyvinylacetate phthalate copolymer, styrene and maleic acid copolymers, polyacrylic acid derivatives such as acrylic acid and acrylic ester
- Some preferred pH-sensitive polymers include shellac; phthalate derivatives, particularly cellulose acetate phthalate, polyvinylacetate phthalate, and hydroxypropylmethylcellulose phthalate; polyacrylic acid derivatives, particularly polymethyl methacrylate blended with acrylic acid and acrylic ester copolymers; and vinyl acetate and crotonic acid copolymers.
- Cellulose acetate phthalate may be applied to latrepirdine tablets to provide delayed release of latrepirdine until the latrepirdine-containing tablet has passed the sensitive duodenal region, that is to delay the release of latrepirdine in the gastrointestinal tract until about 15 minutes, and preferably about 30 minutes, after the latrepirdine-containing tablet has passed from the stomach to the duodenum.
- the CAP coating solution may also contain one or more plasticizers, such as diethyl phthalate, polyethyleneglycol-400, triacetin, triacetin citrate, propylene glycol, and others as known in the art. Some plasticizers are diethyl phthalate and triacetin.
- the CAP coating formulation may also contain one or more emulsifiers, such as polysorbate-80.
- Anionic acrylic copolymers of methacrylic acid and methylmethacrylate are also particularly useful coating materials for delaying the release of latrepirdine from latrepirdine-containing tablets until the tablets have moved to a position in the small intestine which is distal to the duodenum.
- Copolymers of this type are available from RohmPharma Corp, under the trade names Eudragit-L.RTM. and Eudragit-S.RTM, Eudragit-FS.
- Eudragit-L.RTM., Eudragit-S.RTM. and Eudragit FS are anionic copolymers of methacrylic acid and methylmethacrylate.
- the ratio of free carboxyl groups to the esters is approximately 1 :1 in Eudragit-L.RTM. and approximately 1 :2 in Eudragit-S.RTM.. Mixtures of the Eudragits may also be used.
- these acrylic coating polymers may be dissolved in an organic solvent or mixture of organic solvents or may be applied as aqueous colloidal suspensions.
- Useful solvents for the organic coating purpose are acetone, isopropyl alcohol, and methylene chloride. It is generally advisable to include 5-20% plasticizer in coating formulations of acrylic copolymers.
- Useful plasticizers are polyethylene glycols, propylene glycols, diethyl phthalate, dibutyl phthalate, castor oil, and triacetin.
- the delay time before release of latrepirdine, after the "pH-dependent coated tablet” dosage form has exited the stomach, may be controlled by the choice and the relative amounts of the Eudragits used in the coating.
- Eudragit-L.RTM. films dissolve above pH 6.0
- Eudragit-S.RTM. films dissolve above 7.0
- Eudragit FS dissolves above 7.0
- mixtures dissolve at intermediate pH values. Since the pH of the duodenum is approximately 6.0 and the pH of the colon is approximately 7.0, coatings composed of mixtures of Eudragit-L.RTM. and Eudragit-S.RTM. provide protection of the duodenum from latrepirdine.
- Eudragit-S.RTM. or Eudragit FS may be used as the coating material, as described by Dew et al (Br. J. Clin. Pharmac. 14 (1982) 405-408) and Cole et al (Int. J. Pharm., 231 (2002) 83-95).
- preferred coatings comprise from about 9:1 to about 1 :9 Eudragit-L.RTM./Eudragit-S.RTM., more preferably from about 9:1 to about 1 :4 Eudragit-L.RTM./Eudragit-S.RTM..
- the coating may comprise from about 3% to about 70% of the weight of the uncoated tablet core.
- the coating comprises from about 5% to about 50% of the weight of the tablet core.
- a "pH-dependent coated bead” beads (about 0.5 to 3.0 mm in diameter) comprising latrepirdine plus carrier are coated with one or more of the aforementioned pH-sensitive polymers.
- the coated beads may be placed in a capsule or may be compressed into a tablet, with care taken to avoid damaging the polymeric coat on individual beads during tablet compression.
- Preferred coated beads are those which exhibit substantially no release of latrepirdine from the dosage form until the beads have exited the stomach and have resided in the small intestine for about 15 minutes or greater, preferably about 30 minutes or greater, thus assuring that minimal latrepirdine is released in the duodenum.
- Mixtures of a pH- sensitive polymer with a water-insoluble polymer are also included.
- latrepirdine-containing beads may be coated with mixtures of polymers whose solubilities vary at different pH's.
- preferred coatings comprise from about 9:1 to about 1 :9 Eudragit-L.RTM./Eudragit-S.RTM., more preferably from 9:1 to 1 :4 Eudragit-L.RTM./Eudragit-S.RTM..
- the coating may comprise from about 5% to about 200% of the weight of the uncoated bead core.
- the coating comprises from about 10% to about 100% of the weight of the bead core.
- small latrepirdine- containing particles (about 0.01 to 0.5 mm in diameter, preferably 0.05 to 0.5 mm in diameter) are coated with one or more of the aforementioned pH-sensitive polymers.
- the coated particles may be placed in a capsule or may be compressed into a tablet, with care taken to avoid damaging the polymeric coat on individual particles during tablet compression.
- Preferred coated particles are those which exhibit substantially no release of latrepirdine from the dosage form until the particles have exited the stomach and have resided in the small intestine for about 30 minutes or greater, preferably 60 minutes or greater more preferably 90 minutes or greater, thus assuring that minimal latrepirdine is released in the duodenum and upper part of the small intestine.
- Latrepirdine-containing particles are coated with an amount of polymer comprising about 25% to about 200% of the weight of the uncoated latrepirdine-containing particle core.
- a further embodiment constitutes a modification of the pH-dependent coated tablet, pH-dependent coated bead, and pH-dependent coated particle embodiments.
- the latrepirdine-containing core tablet, bead, or particle is first coated with a barrier coat, and then is coated with the pH-dependent coat.
- the function of the barrier coat is to separate latrepirdine from the pH-dependent coat.
- Suitable barrier coatings are composed of water-soluble materials such as sugars such as sucrose, or water- soluble polymers such as hydroxypropyl cellulose, hydroxypropyl methylcellulose, and the like. Hydroxypropyl cellulose and hydroxypropylmethylcellulose are preferred.
- the barrier coat may comprise from about 1 % to about 15%, preferably from about 2% to about 10%, of the weight of the uncoated latrepirdine-containing tablet, bead or particle core.
- latrepirdine-containing tablets, beads and particles may be carried out using equipment known in the art.
- latrepirdine-containing tablet cores may be coated with a pan-coater, such as a Hi-Coater (Freund Corp.), or an Accela-Cota (Manesty Corp., Liverpool).
- Latrepirdine-containing beads and particles are preferably coated using a fluidized bed coater, such as a Wurster coater, utilizing coating equipment available for example from the Glatt Corporation (Ramsey, N.J.).
- Beads may also be coated using a rotary granulator, such as a CF-granulator available from Freund Corp.
- latrepirdine is incorporated in an osmotic bursting device which comprises a tablet core or bead core containing latrepirdine and, optionally, one or more osmagents.
- osmagents are sugars such as glucose, sucrose, mannitol, lactose, and the like; and salts such as sodium chloride, potassium chloride, sodium carbonate, and the like; water-soluble acids such as tartaric acid, fumaric acid, and the like.
- the latrepirdine-containing tablet core or bead core is coated with a polymer which forms a semipermeable membrane, that is, a membrane which is permeable to water but is substantially impermeable to latrepirdine.
- a polymer which forms a semipermeable membrane are cellulose acetate, cellulose acetate butyrate, and ethylcellulose, preferably cellulose acetate.
- the semipermeable coating membrane may alternatively be composed of one or more waxes, such as insect and animal waxes such as beeswax, and vegetable waxes such as carnauba wax and hydrogenated vegetable oils.
- a melt mixture of a polyethylene glycol, e.g., polyethylene glycol-6000, and a hydrogenated oil, e.g., hydrogenated castor oil, may be used as a coating, as described for isoniazid tablets by Yoshino (Capsugel Symposia Series; Current Status on Targeted Drug Delivery to the Gastrointestinal Tract; 1993; pp.185-190).
- Some preferred semipermeable coating materials are cellulose esters and cellulose ethers, polyacrylic acid derivatives such as polyacrylates and polyacrylate esters, and polyvinyl alcohols and polyalkenes such as ethylene vinyl alcohol copolymer.
- Other semipermeable coating materials are cellulose acetate and cellulose acetate butyrate.
- a coated tablet or bead of the "bursting osmotic core" embodiment of this invention When a coated tablet or bead of the "bursting osmotic core" embodiment of this invention is placed in an aqueous environment of use, water passes through the semipermeable membrane into the core, dissolving a portion of the latrepirdine and osmagent, generating a colloidal osmotic pressure which results in bursting of the semipermeable membrane and release of latrepirdine into the aqueous environment.
- the time lag between placement of the dosage form into the aqueous environment of use and release of the enclosed latrepirdine may be chosen.
- Osmotic-bursting devices of this invention are those which exhibit substantially no release of latrepirdine from the dosage form until the dosage form has exited the stomach and has resided in the small intestine for about 15 minutes or greater. Some osmotic-bursting devices exhibit substantially no release of latrepirdine from the dosage form until the dosage form has exited the stomach and has resided in the small intestine for about 30 minutes or greater.
- osmotic-bursting devices exhibit substantially no release of latrepirdine from the dosage form until the dosage form has exited the stomach and has resided in the small intestine for about 90 minutes or greater. Still other osmotic- bursting devices exhibit substantially no release of latrepirdine from the dosage form until the dosage form has exited the stomach and has resided in the small intestine for and most preferably 3 hours or greater, thus assuring that minimal latrepirdine is released in the duodenum and upper small intestine.
- a bursting osmotic core tablet or bead has a tablet or bead core which may contain from about 10-95% latrepirdine, about 0-60% osmagent, as described above, and about 5-20% other pharmaceutical aids such as binders and lubricants.
- the semipermeable membrane coating on a tablet such as a cellulose acetate coating, is present at a weight corresponding to from about 2% to about 30%, preferably from about 3% to about 10%, of the weight of the tablet core.
- the semipermeable membrane coating on a bead, such as a cellulose acetate coating is present at a weight corresponding to from about 2% to about 80% of the weight of the bead core. In another embodiment, the semipermeable coating on a bead is present at a weight corresponding to from 3% to 30% of the weight of the bead core.
- a bursting osmotic core device possesses no mechanism for "sensing" that the device has exited the stomach and entered the duodenum.
- devices of this type release latrepirdine at a predetermined time after entering an aqueous environment, e.g., after being swallowed.
- indigestible non- disintegrating solids such as the "bursting osmotic core devices" of this invention, are emptied from the stomach during phase III of the Interdigestive Migrating Myoelectric Complex (IMMC), which occurs approximately every 2 hr in the human.
- IMMC Interdigestive Migrating Myoelectric Complex
- a bursting osmotic core device may exit the stomach almost immediately after dosing, or as long as 2 hr after dosing.
- indigestible non-disintegrating solids which are ⁇ 1 1 mm in diameter, will empty slowly from the stomach with the contents of the meal (Khosla and Davis, Int. J. Pharmaceut. 62 (1990) R9-R1 1 ).
- the indigestible non-disintegrating solid is greater than about 1 1 mm in diameter, e.g., about the size of a typical tablet, it will be retained in the stomach for the duration of the digestion of the meal, and will exit into the duodenum during phase III of an IMMC, after the entire meal has been digested and has exited the stomach.
- the release of latrepirdine can be delayed until about 15 min or more.
- the release of latrepirdine can be delayed until 30 minutes or more.
- the release of latrepirdine can be delayed until about 90 minutes or greater.
- the release of latrepirdine can be delayed until about 3 hours or greater after the dosage form has exited the stomach.
- a bursting osmotic core device which releases latrepirdine about 3 hr or more after ingestion will show a decrease in metabolism as measured by a reduction of metabolic markers such as A me t.
- a bursting osmotic core device starts to release latrepirdine at about 2.5 hr after entering an aqueous environment, e.g., after ingestion, to more reliably assure that the device releases its latrepirdine distal to the duodenum, when dosed in the fasted state.
- Another "bursting osmotic core device” will start to release latrepirdine at about 4 hr after entering an aqueous environment.
- This 4 hr delay permits dosing in the fed state, and allows for an about 3.5 hr retention in the fed stomach, followed by an approximately 30 minute delay after the dosage form has exited from the stomach. In this way, the release of latrepirdine into the most sensitive portion of the gastrointestinal tract, the duodenum, is minimized.
- a "bursting coated swelling core" a latrepirdine- containing tablet or bead is prepared which also comprises 25-70% of a swellable material, such as a swellable colloid (e.g., gelatin), as described in Milosovich, U.S. Pat. No. 3,247,066, incorporated herein by reference.
- Swelling core materials are hydrogels, e.g., hydrophilic polymers which take up water and swell, such as polyethylene oxides, polyacrylic acid derivatives such as polymethyl methacrylate, polyacrylamides, polyvinyl alcohol, poly-N-vinyl-2-pyrrolidone, carboxymethylcellulose, starches, and the like.
- Swelling hydrogels for this embodiment include polyethylene oxides, carboxymethylcellulose and croscarmellose sodium.
- the colloid/hydrogel-containing latrepirdine-containing core tablet or bead is coated, at least in part, by a semipermeable membrane.
- examples of polymers which provide a semipermeable membrane are cellulose acetate and cellulose acetate butyrate, and ethylcellulose.
- the semipermeable coating membrane may alternatively be composed of one or more waxes, such as insect and animal waxes such as beeswax, and vegetable waxes such as carnauba wax and hydrogenated vegetable oils.
- a melt mixture of a polyethylene glycol, e.g., polyethylene glycol-6000, and a hydrogenated oil, e.g., hydrogenated castor oil, may be used as a coating, as described for isoniazid tablets by Yoshino (Capsugel Symposia Series; Current Status on Targeted Drug Delivery to the Gastrointestinal Tract; 1993; pp.185-190).
- Some semipermeable coating materials are cellulose esters and cellulose ethers, polyacrylic acid derivatives such as polyacrylates and polyacrylate esters, polyvinyl alcohols and polyalkenes such as ethylene vinyl alcohol copolymer, cellulose acetate and cellulose acetate butyrate.
- Preferred bursting coated swelling core devices of this invention are those which exhibit substantially no release of latrepirdine from the dosage form until the dosage form has exited the stomach and has resided in the small intestine for about 15 minutes or greater, preferably about 30 minutes or greater, thus assuring that minimal latrepirdine is released in the duodenum.
- a bursting coated swelling core tablet or bead has a tablet or bead core which may contain from about 10-70% latrepirdine; about 15-60% swelling material, e.g., hydrogel; about 0-15% optional osmagent; and about 5-20% other pharmaceutical aids such as binders and lubricants.
- the semipermeable membrane coating on a tablet preferably a cellulose acetate coating, is present at a weight corresponding to from about 2% to about 30%, preferably from 3% to 10%, of the weight of the tablet core.
- the semipermeable membrane coating on a bead preferably a cellulose acetate coating, is present at a weight corresponding to from about 2% to about 80%, preferably from 3% to 30%, of the weight of the bead core.
- a bursting coated swelling core device possesses no mechanism for sensing that the device has exited the stomach and entered the duodenum.
- devices of this type release their latrepirdine contents at a predetermined time after entering an aqueous environment, e.g., after being swallowed, as previously discussed for bursting osmotic core devices, and the same consideration and preferences apply to making bursting coated swelling core devices.
- a "pH-triggered osmotic bursting device” latrepirdine is incorporated into a device of the type described in allowed commonly assigned co-pending U.S. Pat. No. 5,358,502, issued Oct. 25, 1994, incorporated herein by reference.
- the device comprises latrepirdine and optionally one or more osmagents, surrounded at least in part by a semipermeable membrane.
- the semipermeable membrane is permeable to water and substantially impermeable to latrepirdine and osmagent.
- Useful osmagents are the same as those described above for bursting osmotic core devices.
- Useful semipermeable membrane materials are the same as those described above for bursting osmotic core devices.
- a pH- trigger means is attached to the semipermeable membrane.
- the pH-trigger means is activated by a pH above 5.0, and triggers the sudden delivery of the latrepirdine.
- the pH-trigger means comprises a membrane or polymer coating which surrounds the semipermeable coating.
- the pH-trigger coating contains a polymer which is substantially impermeable and insoluble in the pH range of the stomach, but becomes permeable and soluble at about the pH of the duodenum, about pH 6.0.
- Exemplary pH-sensitive polymers are polyacrylamides, phthalate derivatives such as acid phthalates of carbohydrates, amylose acetate phthalate, cellulose acetate phthalate, other cellulose ester phthalates, cellulose ether phthalates, hydroxypropylcellulose phthalate, hydroxypropylethylcellulose phthalate, hydroxypropylmethylcellulose phthalate, methylcellulose phthalate, polyvinyl acetate phthalate, polyvinyl acetate hydrogen phthalate, sodium cellulose acetate phthalate, starch acid phthalate, styrene-maleic acid dibutyl phthalate copolymer, styrene- maleic acid polyvinylacetate phthalate copolymer, styrene and maleic acid copolymers, polyacrylic acid derivatives such as acrylic acid and acrylic ester copolymers, polymethacrylic acid and esters thereof, poly acrylic methacrylic acid copolymers, shellac, and vinyl a
- Preferred pH-sensitive polymers include shellac; phthalate derivatives, particularly cellulose acetate phthalate, polyvinylacetate phthalate, and hydroxypropylmethylcellulose phthalate; polyacrylic acid derivatives, particularly polymethyl methacrylate blended with acrylic acid and acrylic ester copolymers; and vinyl acetate and crotonic acid copolymers.
- phthalate derivatives particularly cellulose acetate phthalate, polyvinylacetate phthalate, and hydroxypropylmethylcellulose phthalate
- polyacrylic acid derivatives particularly polymethyl methacrylate blended with acrylic acid and acrylic ester copolymers
- vinyl acetate and crotonic acid copolymers vinyl acetate and crotonic acid copolymers.
- cellulose acetate phthalate is available as a latex under the tradename Aquateric.RTM. (registered trademark of FMC Corp., Philadelphia, Pa.)
- acrylic copolymers are available under the tradenames Eudragit-R.RTM.
- the pH-trigger coating may also comprise a mixture of polymers, for example cellulose acetate and cellulose acetate phthalate.
- Another suitable mixture comprises Eudragit-L.RTM. and Eudragit-S.RTM.; the ratio of the two, and the coating thickness, defining the sensitivity of the "trigger", e.g., the pH at which the outer pH-trigger coating weakens or dissolves.
- a pH-triggered osmotic bursting device generally operates as follows. After oral ingestion, the pH-trigger coating, which surrounds the semipermeable coating, which in turn surrounds the latrepirdine-containing core tablet or bead, remains undissolved and intact in the stomach. In the stomach, water may or may not commence penetration through the pH-trigger coating and the semipermeable coating, thus starting hydration of the core, which contains latrepirdine and optional osmagent. After the device has exited the stomach and has entered the small intestine, the pH-trigger coating rapidly disintegrates and dissolves, and water passes through the semipermeable coating, dissolving latrepirdine and optional osmagent within the core.
- the semipermeable coating fails, and the device bursts, releasing latrepirdine. It is preferred that this bursting and release of latrepirdine occur at about 15 minutes or more, preferably 30 minutes or more, after the pH-triggered osmotic bursting device exits the stomach and enters the duodenum, thus minimizing exposure of the sensitive duodenum to latrepirdine.
- the lag-time or delay-time is controlled by the choice and amount of osmagent in the core, by the choice of semipermeable coating, and by the thickness of the semipermeable coating. It will be appreciated by those skilled in the art, for example, that a thicker semipermeable coating will result in a longer delay after the device has exited the stomach.
- a preferred pH-triggered osmotic bursting device is a bead or tablet core of latrepirdine with optional osmagent, coated with a 3-20% by weight cellulose acetate membrane, coated with a 3-20% by weight membrane composed of about 1 : 1 cellulose ace tat e/cellu lose acetate phthalate.
- Another preferred pH-triggered osmotic bursting device is a bead or tablet core of latrepirdine with optional osmagent, coated with a 3-20% by weight cellulose acetate membrane, coated with a 3-20% by weight membrane comprising from about 9:1 to about 1 : 1 Eudragit-L.RTM./Eudragit-S.RTM.
- a pH-triggered osmotic bursting device possesses a mechanism for sensing that the device has exited the stomach, intersubject variability in gastric emptying is not significant.
- a "pH-triggered bursting coated swelling core” a tablet core or bead containing latrepirdine and a swelling material is coated with a semipermeable coating which is further coated with a pH-sensitive coating.
- the core composition, including choice of swelling material is as described above for the bursting coated swelling core embodiment.
- the choice of semipermeable coating material and pH-sensitive coating material are as described above for the "pH- triggered osmotic core” embodiment.
- a pH-triggered bursting swelling core embodiment generally operates as follows. After oral ingestion, the pH-trigger coating, which surrounds the semipermeable coating, which in turn surrounds the latrepirdine-containing core tablet or bead, remains undissolved and intact in the stomach. In the stomach, water may or may not commence penetration through the pH-trigger coating and the semipermeable coating, thus starting hydration of the core, which contains latrepirdine and water-swellable material, preferably a hydrogel.
- the pH-trigger coating rapidly disintegrates and dissolves, and water passes through the semipermeable coating, dissolving latrepirdine and swelling the water-swellable material within the core.
- the semipermeable coating fails, and the device bursts, releasing latrepirdine.
- This bursting and release of latrepirdine occurs at about 15 minutes or more, around about 30 minutes, after the pH-triggered bursting swelling core device exits the stomach and enters the duodenum, thus minimizing exposure of the sensitive duodenum to latrepirdine.
- a pH-triggered bursting swelling core device contains a bead or tablet core of latrepirdine with synthetic hydrogel, preferably carboxymethylcellulose, coated with a 3-20% by weight cellulose acetate membrane, coated with a 3-20% by weight membrane composed of about 1 : 1 cellulose acetate/cellulose acetate phthalate.
- Another pH-triggered bursting swelling core device contains a bead or tablet core of latrepirdine with synthetic hydrogel, preferably carboxymethylcellulose, coated with a 3-20% by weight cellulose acetate membrane, coated with a 3-20% by weight membrane composed of from about 9: 1 to about 1 :1 Eudragit-L.RTM./Eudragit- S.RTM.
- a pH-triggered bursting swelling core device possesses a mechanism for sensing that the device has exited the stomach, intersubject variability in gastric emptying is not significant.
- Delayed release embodiments of the invention are solid dosage forms for oral administration comprising latrepirdine and a pharmaceutically acceptable carrier, which release not more than 10% of their incorporated latrepirdine into a mammal's stomach, and which release not more than an additional 10% during the first 15 minutes after entering said mammal's duodenum.
- the timing of release of latrepirdine in the stomach or duodenum may be tested utilizing a variety of approaches including, but not limited to, x-ray evaluation, nuclear magnetic resonance imaging, gamma scintigraphy, or direct sampling of the gastric and duodenal contents via intubation. These tests, while possible, can be very difficult to carry out in humans.
- a more convenient test for a delayed release embodiment of the current invention is a two stage in vitro dissolution test which was previously described above as "in vitro dissolution test 2".
- Figure 1 Representative in vitro dissolution profiles for latrepirdine immediate release tablets measured using dissolution test 1 .
- Figure 2 Representative in vitro dissolution profiles for 90 and 200 wt% enteric coated Eudragit FS30 melt spray congeal multiparticulates. Dissolution was measured using dissolution test 2.
- Figure 3 Representative in vitro dissolution profile 200 wt% enteric coated Eudragit L30D-55 melt spray congeal multiparticulates. Dissolution was measured using dissolution test 2.
- Figure 4 Representative in vitro dissolution profiles for 10.5 wt% and 21 wt% AMT 3: 1 CA:HPC membrane coatings. Dissolution was completed by dissolution test 1.
- the dose and strength of the tablets is defined in terms of weight of latrepirdine dihydrochloride (a 5mg tablet is 4.1mg of latrepirdine free base; (a 10 mg tablet is 8.2 mg of latrepirdine free base); (a 20 mg tablet is 16.4 mg of Latrepirdine free base) .
- the API has a latrepirdine dihydrochloride theoretical factor of 91.59% (to correct for the dihydrate).
- the excipients from the above table (microcrystalline cellulose, lactose and sodium starch glycolate) and latrepirdine di-HCI were loaded into an appropriately sized bin, ensuring that the latrepirdine di-HCI is sandwiched between the excipients.
- the premix was blended for 10 minutes at 12 +/- 1 RPM and then passed through a rotary mill equipped with a 1 .0mm screen, running at approximately 1000 RPM.
- the sieved blend was collect in an appropriately sized bin and to this was added intragranular magnesium stearate and blend for 5 minutes at 12 +/- 1 RPM.
- the lubricated blend was processed through a Gerteis roller compactor equipped with an inline oscillated mill and the granules were collected into an appropriately sized bin.
- the targeted ribbon solid fraction was 0.67 (0.65-0.72).
- the collected granules were blended in an appropriately sized bin. To this was added extragranular magnesium stearate and this mixture was blended for 5 minutes at 12 +/- 1 RPM.
- Tablets were compressed on a rotary tablet press to an average target weight of l OO.Omg, a target hardness of 5-9 kP, and a target thickness of 2.4-2.9 mm.
- the tablets were passed through a deduster.
- An aqueous film-coating was applied to the tablets to a target weight gain of 4.0% using a perforated coating pan.
- a 3 kg batch of microspheres was prepared as follows: 1230 g of glyceryl behenate was added to an 8 quart V-Blender and blended for 1 minute at 12 ⁇ 1 RPM to coat the metal surfaces. 540 g of latrepirdine dihydrochloride dihydrate was then added to the center of the blender. Another 1230 g of glyceryl behenate was added to the blender ensuring that the latrepirdine was completely covered. The resulting mixture was blended for 15 minutes at 12 ⁇ 1 RPM. The blended mixture was then discharged into a plastic charge bag and transferred to a K-Tron Soder powder feeder. The powder feeder was calibrated to deliver the blend at a rate of 50 ⁇ 5 g/min.
- the blend was processed through a Leistritz 17mm twin-screw extruder fed by the K-Tron Soder powder feeder and coupled to the melt-spray-congeal (MSC) spinning disk according to the operating conditions in Table 1.
- microspheres were transferred to trays and annealed in a temperature and relative humidity controlled oven at 40°C/75% RH for 24 hours with a maximum bed depth of 0.5 inch. After annealing the microspheres were passed through a 425 micron screen to break up any weak agglomerates formed during annealing.
- 162.9 g of purified water was added to an appropriately sized container and heated on a hot plate while stirring until the water temperature reached 70-75°C. Meanwhile, 4 g of triethyl citrate and 4 g of polysorbate 80 were added to a separate container, and 3.2 g of glyceryl monostearate was weighted out onto a piece of wax paper and set aside. Once the water was heated to 70-75°C it was poured into the container with the triethyl citrate and polysorbate 80. The glyceryl monostearate was added to the water/triethyl citrate/polysorbate 80 mixture while homogenizing on a high-shear mixer at 3500-4000 rpm.
- the overall mixture was homogenized for 10-15 minutes. Another 162.9 g of purified water was added to the homogenized glyceryl monostearate suspension and was mixed with a conventional stirrer until the suspension cooled down below 30°C. 263.2 g of Eudragit® FS30D dispersion was weighed out into a separate container. The cooled glyceryl monostearate suspension was then poured slowly into the Eudragit® FS30D dispersion while mixing gently. The final coating solution was passed through a 425 micron screen just before use.
- the coating was carried out on a Glatt GPCG-1 fluid bed coater with a 3.5" ID product chamber and a Wurster column insert charged with 50 g of core microspheres from Example 1.
- the Wurster column gap was set at 5mm.
- a spray rate of 1 .5 - 2.2 g/min was maintained during the run with a product temperature of 26-28°C at an air flow setting of 0.3 bar.
- the microspheres were sprayed until 300 g of coating solution was deposited, which corresponds to a weight gain of approximately 90%.
- the inlet air heater was then turned off and the microspheres were dried under fluidization for 25 minutes.
- the resulting microspheres were passed through a 425 micron screen and any coated material greater than 425 micron was discarded.
- the sieved microspheres were then blended with 2% Talc by weight in a Turbula bottle blender.
- Microspheres 181.3 g of purified water was added to an appropriately sized container and heated on a hot plate while stirring until the water temperature reached 70-75°C. Meanwhile, 4.4 g of triethyl citrate and 4.4 g of polysorbate 80 were added to a separate container, and 3.6 g of glyceryl monostearate was weighted out onto a piece of wax paper and set aside. Once the water was heated to 70-75°C it was poured into the container with the triethyl citrate and polysorbate 80. The glyceryl monostearate was added to the water/triethyl citrate/polysorbate 80 mixture while homogenizing on a high-shear mixer at 3500-4000 rpm.
- the overall mixture was homogenized for 10-15 minutes. Another 181.3 g of purified water was added to the homogenized glyceryl monostearate suspension and was mixed with a conventional stirrer until the suspension cooled down below 30°C. 293 g of Eudragit® FS30D dispersion was weighed out into a separate container. The cooled glyceryl monostearate suspension was then poured slowly into the Eudragit® FS30D dispersion while mixing gently. The final coating solution was passed through a 425 micron screen just before use.
- the coating was carried out on a Glatt GPCG-1 fluid bed coater with a 3.5" ID product chamber and a Wurster column insert charged with 25 g of core microspheres from Example 1.
- the Wurster column gap was set at 5mm.
- a spray rate of 1 .5 - 2.2 g/min was maintained during the run with a product temperature of 26-28°C at an air flow setting of 0.3 bar.
- the microspheres were sprayed until 334 g of coating solution was deposited, which corresponds to a weight gain of approximately 200%.
- the inlet air heater was then turned off and the microspheres were dried under fluidization for 25 minutes.
- the resulting microspheres were passed through a 425 micron screen and any coated material greater than 425 micron was discarded.
- the sieved microspheres were then blended with 2% Talc by weight in a Turbula bottle blender.
- the overall mixture was homogenized for 10-15 minutes.
- Another 42.2 g of purified water was added to the homogenized glyceryl monostearate suspension and was mixed with a conventional stirrer until the suspension cooled down below 30°C.
- 129 g of Eudragit® L30D-55 dispersion was weighed out into a separate container.
- the cooled glyceryl monostearate suspension was then poured slowly into the Eudragit® L30D-55 dispersion while mixing gently.
- the final coating solution was passed through a 425 micron screen just before use.
- the coating was carried out on a Glatt GPCG-1 fluid bed coater with a 3.5" ID product chamber and a Wurster column insert charged with 25 g of core microspheres from Example 1.
- the Wurster column gap was set at 5mm.
- a spray rate of 1 .2 - 1.6 g/min was maintained during the run with a product temperature of 30-32°C at an air flow setting of 0.3 bar.
- the microspheres were sprayed until 1 10 g of coating solution was deposited, which corresponds to a weight gain of approximately 90%.
- the inlet air heater was then turned off and the microspheres were dried under fluidization for 20 minutes.
- the resulting microspheres were passed through a 425 micron screen and any coated material greater than 425 micron was discarded.
- the overall mixture was homogenized for 10-15 minutes. Another 93.2 g of purified water was added to the homogenized glyceryl monostearate suspension and was mixed with a conventional stirrer until the suspension cooled down below 30°C. 285.2 g of Eudragit® L30D-55 dispersion was weighed out into a separate container. The cooled glyceryl monostearate suspension was then poured slowly into the Eudragit® L30D-55 dispersion while mixing gently. The final coating solution was passed through a 425 micron screen just before use.
- the coating was carried out on a Glatt GPCG-1 fluid bed coater with a 3.5" ID product chamber and a Wurster column insert charged with 25 g of core microspheres from Example 1.
- the Wurster column gap was set at 5mm.
- a spray rate of 1 .3 - 1.8 g/min was maintained during the run with a product temperature of 30-32°C at an air flow setting of 0.3 bar.
- the microspheres were sprayed until 243 g of coating solution was deposited, which corresponds to a weight gain of approximately 200%.
- the inlet air heater was then turned off and the microspheres were dried under fluidization for 20 minutes.
- the resulting microspheres were passed through a 425 micron screen and any coated material greater than 425 micron was discarded.
- a drug layer solution is prepared by adding 1246.67g purified water to a container equipped with a mixer. With vigorous mixing, 20. Og hydroxypropyl methylcellulose (Methocel E5), and 200. OOg latrepirdine hydrochloride are dissolved therein. Mixing is continued until complete dissolution is achieved.
- Non-pareil seeds (1 OOOg, 20/25 mesh,) are dispensed into a Wurster fluid bed coater (Glatt 1 .1 ). After fluidization of the non-pareils, spraying of the drug layer solution is commenced to layer drug solution effectively onto the seeds. Spraying is continued until the drug layer solution is exhausted (approximately 22% w/w weight gain). The beads are then sieved through a 710 ⁇ screen and an 850 ⁇ to remove any agglomerated or broken beads. An isolation layer solution is prepared by adding 664.0 g of water to a container equipped with a mixer. With vigorous mixing, 50. Og hydroxypropyl methylcellulose (methocel E5) is mixed until a complete dissolution is achieved.
- Methocel E5 hydroxypropyl methylcellulose
- the drug layer cores are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidization of the drug layered cores, spraying of the isolation layer is commenced to layer the isolation solution onto the cores. Spraying is continued until all the solution is exhausted (approximately a 5% w/w weight gain).
- the beads are dried under the same conditions for 5 minutes. The beads are then sieved through a 710 ⁇ screen and an 850 ⁇ screen to remove any agglomerated or broken beads.
- the beads are to be transferred to trays and annealed in a temperature and relative humidity controlled oven at 40°C/75% RH for at least 24 hours.
- a drug layer solution is prepared by adding 1246.67g purified water to a container equipped with a mixer. With vigorous mixing, 20. Og hydroxypropyl methylcellulose (Methocel E5), and 200. OOg latrepirdine hydrochloride are dissolved therein. Mixing is continued until complete dissolution is achieved.
- Microcrystalline Cellulose (MCC) seeds 1000g, 20/25 mesh, are dispensed into a Wurster fluid bed coater (Glatt 1 .1 ). After fluidization of the MCC seeds, spraying of the drug layer solution is commenced to layer drug solution effectively onto the seeds. Spraying is continued until the drug layer solution is exhausted (approximately 22% w/w weight gain). The beads are then sieved through a 710 ⁇ screen and an 850 ⁇ to remove any agglomerated or broken beads.
- MCC Microcrystalline Cellulose
- An isolation layer solution is prepared by adding 664.0 g of water to a container equipped with a mixer. With vigorous mixing, 50. Og hydroxypropyl methylcellulose (methocel E5) is mixed until a complete dissolution is achieved.
- the drug layer cores are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidization of the drug layered cores, spraying of the isolation layer is commenced to layer the isolation solution onto the cores. Spraying is continued until all the solution is exhausted (approximately a 5% w/w weight gain).
- the beads are dried under the same conditions for 5 minutes. The beads are then sieved through a 710 ⁇ screen and an 850 ⁇ screen to remove any agglomerated or broken beads. The beads are to be transferred to trays and annealed in a temperature and relative humidity controlled oven at 40°C/75% RH for at least 24 hours.
- a controlled release layer is prepared by adding 60 g of water to a container equipped with a mixer and heating to 70-80°C With vigorous mixing, add 6.13g of triethyl citrate, 7.60 g of glyceryl monostearate and 7.60g of Polysorbate 80 (as 33% aqueous solution). Add 144.60g of water to a container equipped with a mixer and with mixing add the emulsion previously prepared with mixing and allow to cool to room temperature. Add this dispersion to 279.33g of Methacrylic Acid - Ethyl Acrylate Copolymer (1 : 1 ) Dispersion 30 Per Cent (Eudragit® L30-D55) in a suitable container with a mixer and stir gently. Prior to coating pour the coating suspension through a 0.5 mm screen to remove any large agglomerates.
- the drug layer cores as described in example 1 or 2 are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidization of the drug layered cores, spraying of the controlled release layer suspension is commenced to layer the controlled release suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 10% w/w weight gain).
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through a 850 ⁇ screen and an 1000 ⁇ screen to remove any agglomerated or broken beads.
- a controlled release layer is prepared by adding 90 g of water to a container equipped with a mixer and heating to 70-80°C With vigorous mixing, add 10.67g of triethyl citrate, 13.33 g of glyceryl monostearate and 13.33g of Polysorbate 80 (as 33% aqueous solution). Add 268.07g of water to a container equipped with a mixer and with mixing add the emulsion previously prepared with mixing and allow to cool to room temperature. Add this dispersion to 488.87g of Methacrylic Acid - Ethyl Acrylate Copolymer (1 : 1 ) Dispersion 30 Per Cent (Eudragit® L30-D55) in a suitable container with a mixer and stir gently. Prior to coating pour the coating suspension through a 0.5 mm screen to remove any large agglomerates.
- the drug layer cores as described in Example 6 are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidization of the drug layered cores, spraying of the controlled release layer suspension is commenced to layer the controlled release suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 17.5% w/w weight gain).
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through a 850 ⁇ screen and an 1000 ⁇ screen to remove any agglomerated or broken beads.
- a controlled release layer is prepared by adding 150 g of water to a container equipped with a mixer and heating to 70-80°C With vigorous mixing, add 15.27g of triethyl citrate, 19.0 g of glyceryl monostearate and 19.0g of Polysorbate 80 (as 33% aqueous solution). Add 361.53g of water to a container equipped with a mixer and with mixing add the emulsion previously prepared to Methacrylic Acid - Ethyl Acrylate Copolymer (1 : 1 ) Dispersion 30 Per Cent (Eudragit® L30-D55) in a suitable container with a mixer and stir gently. Prior to coating pour the coating suspension through a 0.5 mm screen to remove any large agglomerates.
- the drug layer cores as described in Example 6 or 7 are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidization of the drug layered cores, spraying of the controlled release layer is commenced to layer the controlled release suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 25% w/w weight gain).
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through a 850 ⁇ screen and an 1000 ⁇ screen to remove any agglomerated or broken beads.
- a controlled release layer is prepared by adding 60 g of water to a container equipped with a mixer and heating to 70-80°C With vigorous mixing, add 6.13g of triethyl citrate, 7.60 g of glyceryl monostearate and 7.60g of Polysorbate 80 (as 33% aqueous solution). Add 144.60g of water to a container equipped with a mixer and with mixing add the emulsion previously prepared with mixing and allow to cool to room temperature. Add this dispersion to 279.33g of Methacrylic Acid - Ethyl Acrylate Copolymer (1 : 1 ) Dispersion 30 Per Cent (Eudragit® FS30 D) in a suitable container with a mixer and stir gently. Prior to coating pour the coating suspension through a 0.5 mm screen to remove any large agglomerates.
- the drug layer cores as described in Example 6 or 7 are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidization of the drug layered cores, spraying of the controlled release layer suspension is commenced to layer the controlled release suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 10% w/w weight gain).
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through a 850 ⁇ screen and an 1000 ⁇ screen to remove any agglomerated or broken beads.
- a controlled release layer is prepared by adding 90 g of water to a container equipped with a mixer and heating to 70-80°C With vigorous mixing, add 10.67g of triethyl citrate, 13.33 g of glyceryl monostearate and 13.33g of Polysorbate 80 (as 33% aqueous solution). Add 268.07g of water to a container equipped with a mixer and with mixing add the emulsion previously prepared with mixing and allow to cool to room temperature. Add this dispersion to 488.87g of Methacrylic Acid - Ethyl Acrylate Copolymer (1 :1 ) Dispersion 30 Per Cent (Eudragit® FS30 D) in a suitable container with a mixer and stir gently.
- the drug layer cores as described in example 6 or 7 are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidization of the drug layered cores, spraying of the controlled release layer suspension is commenced to layer the controlled release suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 17.5% w/w weight gain).
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through a 850 ⁇ screen and an 1000 ⁇ screen to remove any agglomerated or broken beads.
- a controlled release layer is prepared by adding 150 g of water to a container equipped with a mixer and heating to 70-80°C With vigorous mixing, add 15.27g of triethyl citrate, 19.0 g of glyceryl monostearate and 19.0g of Polysorbate 80 (as 33% aqueous solution). Add 361.53g of water to a container equipped with a mixer and with mixing add the emulsion previously prepared to Methacrylic Acid - Ethyl Acrylate Copolymer (1 : 1 ) Dispersion 30 Per Cent (Eudragit® FS30 D) in a suitable container with a mixer and stir gently. Prior to coating pour the coating suspension through a 0.5 mm screen to remove any large agglomerates.
- the drug layer cores as described in example 6 or 7 are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidization of the drug layered cores, spraying of the controlled release layer is commenced to layer the controlled release suspension onto the cores. Spraying is continued until all the solution is exhausted (approximately a 25% w/w weight gain).
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through a 850 ⁇ screen and an 1000 ⁇ screen to remove any agglomerated or broken beads.
- the metal surfaces of a 25L bin were pre-coated by adding the batch quantity (also see Table 2 below), 2199.20g, of Avicel (PH 102) and blending for 1 minute at 12 +/- 1 RPM.
- the Fumaric acid, 800g, and one-half the batch quantity of mannitol, 2198.8g, were added to a 25L sized plastic charge bag and gently mixed together by inverting the bag.
- the 523.2g of latrepirdine 2HCI was added to the center of the bin, meanwhile saving the container it was weighed in*.
- the latrepirdine 2HCI container was rinsed twice with portions of the remaining mannitol, which was subsequently added to the bin.
- the contents of the plastic charge bag were emptied into the bin, covering the latrepirdine 2HCI.
- the plastic charge bag was saved. All of the components were blended in the bin for 10 minutes at 12 +/- 1 RPM.
- the blend was passed through a Comil rotary mill equipped with screen and a round edge impeller running at approximately 750 RPM.
- the blend was collected in the saved plastic charge bag.
- the bin and mill were flushed to remove any residual API by adding the remaining mannitol to the bin, blending for 1 minute at 12 +/- 1 RPM, then releasing contents through the mill into the plastic charge bag.
- the milled blend was then transferred back to the bin blender.
- the plastic charge bag was saved.
- the bin contents were blended for 10 minutes at 12 +/- 1 RPM.
- Intragranular magnesium stearate, 40g** was added to the bin and blended for 5 minutes at 12 +/- 1 RPM.
- the lubricated blend was processed through a Gerteis roller compactor equipped with knurled rollers, side rims, and an inline oscillating mill containing a pocket rotor and a 0.8mm rasping plate.
- the target ribbon solid fraction was 0.71 (0.70-0.75) and granules were collected in the saved plastic charge bag.
- Latrepirdine di-hydrochloride, 250mg (15mgA) tablet cores were coated by a functional solvent based film coating spraying process using a Vector LDCS-5 pan coater at small scale (1 kg) with an osmotic polymeric film coating with cellulose acetate as the polymer and hydroxypropyl cellulose as the plasticizer.
- the acetone and water are the co-solvents uses to dissolve the solids and facilitate pore formation.
- the coating solution composition contains 8% solids by weight, comprised of 6% cellulose acetate and 2% hydroxypropyl cellulose (3: 1 ratio) dissolved in 69% acetone and 23% water (3:1 ratio).
- the final dry target of 10.5 wt% coating will be -26 mg of dried coating (75% CA & 25% HPC) per tablet core (on a 250 mg core) to obtain 80% released in 8 hours by in vitro dissolution test 1 .
- the AMT coating was prepared as a 8% (w/w) solution.
- the coating solution was applied to the tablet cores using a Vector LDCS-5 pan coater using the following parameters:
- Latrepirdine di-hydrochloride, 250mg (15mgA) tablet cores were coated by a functional solvent based film coating spraying process using a Vector LDCS-5 pan coater at small scale (1 kg) with an osmotic polymeric film coating with cellulose acetate as the polymer and hydroxypropyl cellulose as the plasticizer.
- the acetone and water are the co-solvents uses to dissolve the solids and facilitate pore formation.
- the coating solution composition contains 8% solids by weight, comprised of 6% cellulose acetate and 2% hydroxypropyl cellulose (3: 1 ratio) dissolved in 69% acetone and 23% water (3:1 ratio).
- the final dry target of 21 wt% coating will be -52.5 mg of dried coating (75% CA & 25% HPC) per tablet core (on a 250 mg core) to obtain 80% released in 12 hours by in vitro dissolution test 1.
- the AMT coating was prepared as a 8% (w/w) solution.
- the coating solution was applied to the tablet cores using a Vector LDCS-5 pan coater using the following parameters: Table 6. Coating parameters used for AMT coating in the Vector LDCS-5 pan
- Latrepirdine di-hydrochloride, 250mg (15mgA) tablet cores were coated by a functional solvent based film coating spraying process using a Vector LDCS-5 pan coater at small scale (1 kg) with an osmotic polymeric film coating with cellulose acetate as the polymer and hydroxypropyl cellulose as the plasticizer.
- the acetone and water are the co-solvents uses to dissolve the solids and facilitate pore formation.
- the coating solution composition contains 5% solids by weight, comprised of 3.5% cellulose acetate and 1 .5% hydroxypropyl cellulose (2.3:1 ratio) dissolved in 85.5% acetone and 9.5% water (9: 1 ratio).
- the final dry target of 8 wt% coating will be -20 mg of dried coating per tablet core (on a 250 mg core) to obtain the specified 80% released in 14 hours by USP dissolution in 270 mOsm SIF at pH 6.8 (simulated intestinal fluid).
- the coated tablets are tray dried for 16 hours at 40°C/30%RH to remove residual solvent.
- a single 900 um (0.9 mm) delivery orifice is then drilled through one side of the semi-permeable membrane in the center of the tablet face (does not matter which face of the tablet) which was completed by hand drilling.
- the membrane coating was prepared as a 5% (w/w) solution.
- the coating solution was applied to the tablet cores using a Vector LDCS-5 pan coater using the following parameters: Table 8. Coating parameters used for AMT coating in the Vector LDCS-5 pan
- Latrepirdine di-hydrochloride, 250mg (15mgA) tablet cores were coated by a functional solvent based film coating spraying process using a Vector LDCS-5 pan coater at small scale (1 kg) with an osmotic polymeric film coating with cellulose acetate as the polymer and hydroxypropyl cellulose as the plasticizer.
- the acetone and water are the co-solvents uses to dissolve the solids and facilitate pore formation.
- the coating solution composition contains 5% solids by weight, comprised of 3.75% cellulose acetate and 1.25% hydroxypropyl cellulose (3:1 ratio) dissolved in 85.5% acetone and 9.5% water.
- the final dry target of 12 wt% coating will be -30 mg of dried coating per tablet core (on a 250 mg core) to obtain the specified 80% released in 24 hours by USP dissolution in 270 mOsm SIF at pH 6.8 (simulated intestinal fluid).
- the coated tablets are tray dried for 16 hours at 40°C/30%RH to remove residual solvent.
- a single 900 um (0.9 mm) delivery orifice is then drilled through one side of the semi-permeable membrane in the center of the tablet face (does not matter which face of the tablet) which was completed by hand drilling.
- the membrane coating was prepared as a 5% (w/w) solution.
- the coating solution was applied to the tablet cores using a Vector LDCS-5 pan coater using the following parameters:
- the metal surfaces of a 25L bin are pre-coated by adding the batch quantity (also see Table 11 below), 2968g, of Avicel (PH 102) and blending for 1 minute at 12 +/- 1 RPM.
- One-half the batch quantity of Methocel, 1800g, and the Cabosil, 40g, is added to a 25L sized plastic charge bag and gently mixed together by inverting the bag.
- the 1312g of latrepirdine 2HCI is added to the center of the bin, meanwhile saving the container it is weighed in.
- the latrepirdine 2HCI container is rinsed twice with portions of the remaining Methocel, which is subsequently added to the bin.
- the contents of the plastic charge bag are emptied into the bin, covering the latrepirdine 2HCI.
- the plastic charge bag is saved. All of the components are blended in the bin for 10 minutes at 12 +/- 1 RPM.
- the blend is passed through a Comil rotary mill equipped with a 0.55" screen and a round edge impeller running at approximately 750 RPM.
- the blend is collected in the saved plastic charge bag.
- the bin and mill are flushed to remove any residual API by adding the remaining mannitol to the bin, blending for 1 minute at 12 +/- 1 RPM, then releasing contents through the mill into the plastic charge bag.
- the milled blend is then transferred back to the bin blender.
- the plastic charge bag is saved.
- the bin contents are blended for 10 minutes at 12 +/- 1 RPM.
- Intragranular magnesium stearate 40g**, is added to the bin and blended for 5 minutes at 12 +/- 1 RPM.
- the lubricated blend is processed through a Gerteis roller compactor equipped with knurled rollers, side rims, and an inline oscillating mill containing a pocket rotor and a 0.8mm rasping plate.
- the target ribbon solid fraction is 0.71 (0.70-0.75) and granules are collected in the saved plastic charge bag.
- the collected granules are transferred back to the bin blender and blended for 10 minutes at 12 +/- 1 RPM.
- Extragranular magnesium stearate, 40g** is added to the bin and contents are blended for 5 minutes at 12 +/- 1 RPM.
- Final blend is affixed above a Kilian T-100 rotary tablet press. Tablets are compressed using 13/32" SRC tooling, to an average target weight of 400 mg +/- 5% and a target hardness of 7 kP. Tablets are passed through a deduster and a metal detector.
- the lubricated blend is processed through a roller compactor equipped with knurled rollers, side rims, and an inline oscillating mill containing a pocket rotor and a 0.8mm rasping plate.
- the target ribbon solid fraction is 0.7-0.75.
- the collected granules are weighed to calculate the correct amount of extragrannular magnesium sterate which is then weighed out.
- the granules are transferred back to the bin blender and blended for 10 minutes at 12 +/- 1 RPM.
- the extragranular magnesium stearate is added to the bin and contents are blended for 5 minutes at 12 +/- 1 RPM.
- Latrepirdine SCT swell layer composition swell layer target weight for 60 mg active dose or 330 mg active layer total weight is 165 mg.
- the drug layer and sweller layer are co-compressed using a bi-layer rotary press. Place the drug layer and sweller layer into the two hoppers of a suitable rotary bilayer tablet press, using the drug layer as the first filled layer.
- the press should be set up with 13/32" standard round concave (SRC) tooling. Compress the drug layer only until the desired fill weight (330 mg for a 60 mg dose) is achieved. Compress the drug layer and sweller layer to target weight of 495 mg. The compression of the active layer to swell layer is completed such that the ratio of the two layers is 2: 1 respectively.
- the target first layer hardness value is ⁇ 1 kp, target overall tablet hardness is 6-8 kp.
- Latrepirdine di-hydrochloride, 495 mg (60mgA) tablet cores are coated by a functional solvent based film coating spraying process using a Vector LDCS-5 pan coater at small scale (1 kg) with an osmotic polymeric film coating with cellulose acetate as the polymer and polyethylene glycol 3350 as the plasticizer.
- the acetone and water are the co-solvents uses to dissolve the solids and facilitate pore formation.
- the coating solution composition contains 5% solids by weight, comprised of 4.5% cellulose acetate and 0.5% polyethylene glycol 3350 (9: 1 ratio) dissolved in 85.5% acetone and 9.5% water (9: 1 ratio; see Table 14).
- the final dry target of 22 wt% coating will be -108.9 mg of dried coating per tablet core (on a 495 mg core) to obtain the specified 80% released in 14 hours by USP dissolution in 270 mOsm SIF at pH 6.8 (simulated intestinal fluid). See Table 15 for the coating conditions.
- the coated tablets are tray dried for 16 hours at 40°C/30%RH to remove residual solvent.
- a single 900 um (0.9 mm) delivery orifice is then drilled through the drug side of the bilayer tablet (the side which does not contain colorant). The delivery orifice can be drilled by hand or by laser drilling. Table 14.
- the membrane coating is prepared as a 5% (w/w) solution.
- the coating solution is applied to the tablet cores using a Vector LDCS-5 pan coater using the following parameters:
- a drug layer solution is prepared by adding 1246.67g purified water to a container equipped with a mixer. With vigorous mixing, 20. Og hydroxypropyl methylcellulose (Methocel E5), and 200. OOg latrepirdine hydrochloride are dissolved therein. Mixing is continued until complete dissolution is achieved.
- Microcrystalline Cellulose (MCC) seeds 1000g, 20/25 mesh, are dispensed into a Wurster fluid bed coater (Glatt 1 .1 ). After fluidisation of the MCC seeds, spraying of the drug layer solution is commenced to layer drug solution effectively onto the seeds. Spraying is continued until the drug layer solution is exhausted (approximately 22% w/w weight gain). The beads are then sieved through a 710 ⁇ screen and an 850 ⁇ screen to remove any agglomerated or broken beads.
- An isolation layer solution is prepared by adding 664.0 g of water to a container equipped with a mixer. With vigorous mixing, 50. Og hydroxypropyl methylcellulose (methocel E5) is mixed until a complete solution is achieved.
- the drug layer cores are dispersed into a Wurster fluid bed coater (Glatt 1.1 ) after fluidisation of the drug layered cores, spraying of the isolation layer is commenced to layer the isolation solution onto the cores. Spraying is continued until all the solution is exhausted (approximately a 5% w/w weight gain).
- the beads are dried under the same conditions for 5 minutes. The beads are then sieved through a 710 ⁇ screen and an 850 ⁇ screen to remove any agglomerated or broken beads.
- a controlled release layer is prepared by 1461 .43g of ethanol to a container equipped with a mixer. With vigorous mixing, 10. Og of trethyl citrate and 100g of methacrylic acid co-polymer type A (Eudragit L100-55) is mixed until a complete solution is achieved.
- the drug layer cores are dispersed into a Wuster fluid bed coater (Glatt1.1 ) after fluidisation of the drug layer beads, spraying of the controlled release layer is commenced to layer the controlled release solution on to the beads. Spraying is continued until all the solution is exhausted (approximately 1 1 % w/w weight gain).
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through a 710 ⁇ screen and an 850 ⁇ screen to remove any agglomerated or broken beads.
- the beads are to be transferred to trays and annealed in a temperature and relative humidity controlled oven at 40°C/75% RH for at least 24 hours.
- Pulsatile Beads Drug layered Bead
- a drug layer solution is prepared by adding 1246.67g purified water to a container equipped with a mixer. With vigorous mixing, 20. Og hydroxypropyl methylcellulose (Methocel E5), and 200. OOg latrepirdine hydrochloride are dissolved therein. Mixing is continued until complete dissolution is achieved.
- Microcrystalline Cellulose (MCC) seeds 1000g, 20/25 mesh, are dispensed into a Wurster fluid bed coater (Glatt 1 .1 ). After fluidisation of the MCC seeds, spraying of the drug layer solution is commenced to layer drug solution effectively onto the seeds. Spraying is continued until the drug layer solution is exhausted (approximately 22% w/w weight gain). The beads are then sieved through a 710 ⁇ screen and an 850 ⁇ screen to remove any agglomerated or broken beads.
- a controlled release layer is prepared by 1461 .43g of ethanol to a container equipped with a mixer. With vigorous mixing, 10. Og of trethyl citrate and 100g of methacrylic acid co-polymer type A (Eudragit L100-55) is mixed until a complete solution is achieved.
- the drug layer cores are dispersed into a Wuster fluid bed coater (Glatt1.1 ) after fluidisation of the drug layer beads, spraying of the controlled release layer is commenced to layer the controlled release solution on to the beads. Spraying is continued until all the solution is exhausted (approximately 1 1 % w/w weight gain). The beads are dried under the same conditions for 10 minutes.
- Wuster fluid bed coater Glatt1.1
- the beads are to be transferred to trays and annealed in a temperature and relative humidity controlled oven at 40°C/75% RH for at least 24 hours.
- a sweller layer suspension is prepared by adding 1708.82 g of Ethanol (96%) to a suitable container with a mixer. With vigorous mixing add 50.00g of povidone (Kollidon® 90F). Mix until solution is complete. Add 300. OOg of croscarmellose sodium while mixing. Continue to mix until a homogenous suspension is formed.
- the drug layer cores as described in Example 21 or example 2 are dispersed into Wurster fluid bed coater (Glatt 1 .1 ) after fluidisation of the drug layered cores, spraying of the sweller layer suspension is commenced to layer the sweller layer suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 35% w/w weight gain).
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850 ⁇ screen and a 1000 ⁇ screen to remove any agglomerated or broken beads.
- the rupture coat suspension is prepared by adding 1594.29g of isopropyl alcohol to a suitable container with a mixer. Add 398.57g of water and mix until solution is complete. Add 120.0g of ethylcellulose (EC4CP) with stirring, continue to mix until the ethylcellulose has dissolved. To the solution add with vigorous mixing 30. Og of magnesium stearate. Stir until a homogenous suspension is formed.
- the sweller layer beads as described in Example 23 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidisation of the drug layered cores, spraying of the rupture coat layer suspension is commenced to layer the rupture layer suspension onto the cores.
- Spraying is continued until all the suspension is exhausted (approximately a 15% w/w weight gain).
- the beads are dried under the same conditions for 10 minutes.
- the beads are then sieved through an 850 ⁇ screen and a 1300 ⁇ screen to remove any agglomerated or broken beads.
- An overcoat suspension is prepared by adding 191 .27g of acetone to a suitable container with a mixer. While mixing 8.60g of water is added. Once the solution is complete, add 12.0g of cellulose acetate (CA398-10) and mix until all the cellulose acetate has dissolved. Once the solution is complete add 3.0g of talc and mix until fully dispersed.
- the rupture layer coated beads are dispersed into Wurster fluid bed coater (Glatt 1 .1 ) after fluidisation of the cores, spraying of the overcoat coat suspension is commenced to layer the suspension onto the cores. Spraying is continued until an approximate weight gain of 1.5%w/w is achieved.
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850 ⁇ screen and a 1300 ⁇ screen to remove any agglomerated or broken beads.
- the rupture coat suspension is prepared by adding 2125.71 g of isopropyl alcohol to a suitable container with a mixer. Add 531.43g of water and mix until solution is complete. Add 160.0g of ethylcellulose (EC4CP) with stirring, continue to mix until the ethylcellulose has dissolved. To the solution add with vigorous mixing 40. Og of magnesium stearate. Stir until a homogenous suspension is formed.
- EC4CP ethylcellulose
- the sweller layer beads as described in Example 23 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidisation of the drug layered cores, spraying of the rupture coat layer suspension is commenced to layer the rupture layer suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 20% w/w weight gain).
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850 ⁇ screen and a 1300 ⁇ screen to remove any agglomerated or broken beads.
- An overcoat suspension is prepared by adding 191 .27g of acetone to a suitable container with a mixer. While mixing 8.60g of water is added. Once the solution is complete, add 12.0g of cellulose acetate (CA398-10) and mix until all the cellulose acetate has dissolved. Once the solution is complete add 3.0g of talc and mix until fully dispersed.
- the rupture layer coated beads are dispersed into Wurster fluid bed coater (Glatt 1 .1 ) after fluidisation of the cores, spraying of the overcoat coat suspension is commenced to layer the suspension onto the cores. Spraying is continued until an approximate weight gain of 1.5%w/w is achieved.
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850 ⁇ screen and a 1300 ⁇ screen to remove any agglomerated or broken beads.
- the rupture coat suspension is prepared by adding 2657.14g of isopropyl alcohol to a suitable container with a mixer. Add 664.28g of water and mix until solution is complete. Add 200. Og of ethylcellulose (EC4CP) with stirring, continue to mix until the ethylcellulose has dissolved. To the solution add with vigorous mixing 50. Og of magnesium stearate. Stir until a homogenous suspension is formed.
- EC4CP ethylcellulose
- the sweller layer beads as described in Example 23 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidisation of the drug layered cores, spraying of the rupture coat layer suspension is commenced to layer the rupture layer suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 25% w/w weight gain).
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850 ⁇ screen and a 1300 ⁇ screen to remove any agglomerated or broken beads.
- An overcoat suspension is prepared by adding 191 .27g of acetone to a suitable container with a mixer. While mixing 8.60g of water is added. Once the solution is complete, add 12.0g of cellulose acetate (CA398-10) and mix until all the cellulose acetate has dissolved. Once the solution is complete add 3.0g of talc and mix until fully dispersed.
- the rupture layer coated beads are dispersed into Wurster fluid bed coater (Glatt 1 .1 ) after fluidisation of the cores, spraying of the overcoat coat suspension is commenced to layer the suspension onto the cores. Spraying is continued until an approximate weight gain of 1.5%w/w is achieved.
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850 ⁇ screen and a 1300 ⁇ screen to remove any agglomerated or broken beads.
- the rupture coat suspension is prepared by adding 3188.58g of isopropyl alcohol to a suitable container with a mixer. Add 797.14g of water and mix until solution is complete. Add 240. Og of ethylcellulose (EC4CP) with stirring, continue to mix until the ethylcellulose has dissolved. To the solution add with vigorous mixing 60. Og of magnesium stearate. Stir until a homogenous suspension is formed.
- EC4CP ethylcellulose
- the sweller layer beads as described in Example 23 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidisation of the drug layered cores, spraying of the rupture coat layer suspension is commenced to layer the rupture layer suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 30% w/w weight gain).
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850 ⁇ screen and a 1300 ⁇ screen to remove any agglomerated or broken beads.
- An overcoat suspension is prepared by adding 191 .27g of acetone to a suitable container with a mixer. While mixing 8.60g of water is added. Once the solution is complete, add 12.0g of cellulose acetate (CA398-10) and mix until all the cellulose acetate has dissolved. Once the solution is complete add 3.0g of talc and mix until fully dispersed.
- the rupture layer coated beads are dispersed into Wurster fluid bed coater (Glatt 1 .1 ) after fluidisation of the cores, spraying of the overcoat coat suspension is commenced to layer the suspension onto the cores. Spraying is continued until an approximate weight gain of 1.5%w/w is achieved.
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850 ⁇ screen and a 1300 ⁇ screen to remove any agglomerated or broken beads.
- the rupture coat suspension is prepared by adding 3720. Og of isopropyl alcohol to a suitable container with a mixer. Add 7930. Og of water and mix until solution is complete. Add 280. Og of ethylcellulose (EC4CP) with stirring, continue to mix until the ethylcellulose has dissolved. To the solution add with vigorous mixing 70. Og of magnesium stearate. Stir until a homogenous suspension is formed
- the sweller layer beads as described in Example 23 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidisation of the drug layered cores, spraying of the rupture coat layer suspension is commenced to layer the rupture layer suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 35% w/w weight gain).
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850 ⁇ screen and a 1300 ⁇ screen to remove any agglomerated or broken beads.
- An overcoat suspension is prepared by adding 191.27g of acetone to a suitable container with a mixer. While mixing 8.60g of water is added. Once the solution is complete, add 12.0g of cellulose acetate (CA398-10) and mix until all the cellulose acetate has dissolved. Once the solution is complete add 3.0g of talc and mix until fully dispersed.
- the rupture layer coated beads are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidisation of the cores, spraying of the overcoat coat suspension is commenced to layer the suspension onto the cores. Spraying is continued until an approximate weight gain of 1.5%w/w is achieved.
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850 ⁇ screen and a 1300 ⁇ screen to remove any agglomerated or broken beads.
- the rupture coat suspension is prepared by adding 4251 .42g of isopropyl alcohol to a suitable container with a mixer. Add 1062.86g of water and mix until solution is complete. Add 320. Og of ethylcellulose (EC4CP) with stirring, continue to mix until the ethylcellulose has dissolved. To the solution add with vigorous mixing 80. Og of magnesium stearate. Stir until a homogenous suspension is formed
- the sweller layer beads as described in Example 23 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidisation of the drug layered cores, spraying of the rupture coat layer suspension is commenced to layer the rupture layer suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 40% w/w weight gain).
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850 ⁇ screen and a 1300 ⁇ screen to remove any agglomerated or broken beads.
- An overcoat suspension is prepared by adding 191 .27g of acetone to a suitable container with a mixer. While mixing 8.60g of water is added. Once the solution is complete, add 12.0g of cellulose acetate (CA398-10) and mix until all the cellulose acetate has dissolved. Once the solution is complete add 3.0g of talc and mix until fully dispersed.
- the rupture layer coated beads are dispersed into Wurster fluid bed coater (Glatt 1 .1 ) after fluidisation of the cores, spraying of the overcoat coat suspension is commenced to layer the suspension onto the cores. Spraying is continued until an approximate weight gain of 1.5%w/w is achieved.
- the beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850 ⁇ screen and a 1300 ⁇ screen to remove any agglomerated or broken beads.
- the study was a randomized, open-label, single dose, 5-period, 5-treatment
- Latrepirdine 20 mg Sustained Release dosage forms (SR1 and SR2): The preparation of these dosage forms is described in Examples 1 , 2 and 4 respectively (above).
- Latrepirdine 60 mg, Sustained Release dosage forms (SR3 and SR4): The preparation of these dosage forms is described in Examples 14, 15 and 16 respectively (above). Four latrepirdine 15 mg SR tablets were given to achieve the 60 mg dose.
- PK samples were analyzed using standard validated analytical methods. Dose normalized natural log transformed AUC 0 -24h, AUC in f, AUCi as t and C max were analyzed for latrepirdine and analyte (when applicable) using a mixed effect model with sequence, period and treatment as fixed effects and subject within sequence as a random effect. Estimates of the adjusted mean differences (Test-Reference) and corresponding 90% confidence intervals were obtained from the model. The adjusted mean differences and 90% confidence intervals for the differences was exponentiated to provide estimates of the ratio of adjusted geometric means (Test/Reference) and 90% confidence intervals for the ratios.
- the immediate release control tablet formulation was the Reference treatment and the sustained release formulations were the Test treatments.
- each latrepirdine SR formulation was determined by calculating the individual ratio of the dose normalized mean area under the plasma concentration versus time curve of the latrepirdine sustained release test formulation (i.e. AUC 0 -inf) to the dose normalized mean area under the plasma concentration versus time curve of the latrepirdine immediate release reference formulation (i.e. AUC 0 -inf)- The corresponding individual ratios are then averaged together.
- the relative bioavailability of the four modified release formulations SR1 , SR2, SR3, and SR4, relative to the IR formulation were approximately 79%, 88%, 118% and 123%, respectively.
- AUC 0 -inf, AUCo-24 and AUCi ast ratio for A met were listed and summarized descriptively by treatment as appropriate.
- the ratios of AUC 0 -inf, AUC 0 -24 and AUCi ast were analyzed using a mixed effect model with sequence, period and treatment as fixed effects and subject within sequence as a random effect. Estimates of the adjusted mean differences between the SR and IR formulations, and corresponding 90% confidence intervals was obtained from the model.
- the PK parameters AUCo-24 , AUC inf , AUCi as t, C max , T max , and t V2 were summarized descriptively by treatment and analyte (when applicable).
- AUC inf and C max individual subject parameters were plotted by treatment for each analyte separately (when applicable).
- Concentrations were listed and summarized descriptively by PK sampling time, treatment and analyte (when applicable).
- Individual subject, mean and median profiles of the concentration-time data were plotted by treatment and analyte (when applicable).
- the metal surfaces of a 20L bin were pre-coated by adding the batch quantity (also see Table 16 below), 554.2 g, of Avicel (PH 200) and blending for 1 minute at 12 +/- 1 RPM.
- the Cab-o-sil, 12.5g, and one-half the batch quantity of mannitol, 554.2 g, were added to the 20L bin.
- the 800.0 g of latrepirdine 2HCI was added to the center of the bin after hand sieving through a #30 mesh sieve (600 micron), meanwhile saving the container it was weighed in.
- the latrepirdine 2HCI container was rinsed twice with portions of the remaining mannitol, which was subsequently added to the bin. The remaining mannitol was emptied into the bin, covering the latrepirdine 2HCI. All of the components were blended in the bin for 10 minutes at 25 +/- 1 RPM.
- the blend was passed through a Comil rotary mill equipped with a 0.042" screen and a round edge impeller running at approximately 300 RPM. The blend was collected in the saved plastic charge bag. The milled blend was then transferred back to the bin blender. The bin contents were blended for 10 minutes at 25 +/- 1 RPM.
- Final blend was affixed above a Kilian T-100 rotary tablet press. Tablets were compressed using 5/16" SRC tooling, to an average target weight of 250. Omg +/- 3% and a target hardness of 7.5 kP. Tablets were passed through a deduster.
Abstract
Oral sustained release compositions of latrepirdine for the treatment of neurodegenerative diseases, and especially Alzheimer's disease (AD) and Huntington's disease, are disclosed herein. Also provided are process for their manufacture.
Description
LATREPIRDINE ORAL SUSTAINED RELEASE DOSAGE FORMS
FIELD OF THE INVENTION
The invention provides sustained release formulations comprising latrepirdine or pharmaceutically acceptable salts there of. The formulations have desirable pharmacokinetic characteristics. Examples include AUC, Cmax, dosage-corrected AUC, AUC/Cmax, Ci2 and dosage-corrected Cmax.
BACKGROUND OF THE INVENTION
The present invention relates to oral sustained release compositions of 2,8- dimethyl-5-[2-(6-methylpyridin-3-yl)ethyl]-3,4-dihydro-1 H-pyrido[4,3-b]indole
(hereinafter latrepirdine, also known in the literature as Dimebon) for the treatment of neurodegenerative diseases, and especially Alzheimer's disease (AD) and Huntington's disease.
Latrepirdine is being commercially developed as an immediate release tablet form with doses ranging from 5 mg to 20 mg administered TID (three times a day). While the proposed commercial dosage form provides efficacious blood levels of latrepirdine to subjects, it has been observed in clinical studies that there is a substantial amount of metabolism in most subjects. The major metabolites produced are not considered to be appreciably active relative to the parent, latrepirdine. Additionally, there is a positive food effect seen in the majority of subjects. That is, the systemic absorption of the drug is enhanced when the immediate release tablets are taken with food. It is therefore desired to provide an oral sustained release dosage form containing latrepirdine that overcomes one or more of the disadvantages of an immediate release form. A sustained release dosage form that decreases the dosing frequency, does not undergo the degree of metabolism as the immediate release tablet dosage form, and/or that can be taken with or without food while maintaining similar to identical bioavailability in the fed or fasted state of the subject is particularly desired.
Compared to immediate release formulations, a sustained release formulation containing a physiologically active level of drug allows blood concentrations of the drug to be maintained at or above the therapeutic concentration. Accordingly, by achieving the sustained-release characteristics of a drug it may be possible to reduce the number of dosings while providing the same or better therapeutic effects, thus
potentially improving compliance. With the sustained-release characteristics of the drug, it may also be possible to avoid a rapid increase in blood plasma concentration levels immediately after administration of the drug, thus potentially reducing or eliminating adverse side effects. There is a need in the art for new drug formulations to treat Alzheimer's disease that reduce the dosing frequency which translates to greater compliance or greater efficacy. The invention is directed to these, as well as other, important ends.
SUMMARY OF THE INVENTION
The present invention relates to oral sustained release compositions of latrepirdine for the treatment of neurodegenerative diseases, and especially Alzheimer's disease (AD) and Huntington's disease. Sustained release of latrepirdine may be accomplished by any means known in the pharmaceutical arts, including but not limited to the use of osmotic dosage forms, matrix dosage forms, multiparticulate dosage forms, gastric retentive dosage forms, and pulsatile dosage forms.
2,8-dimethyl-5-[2-(6-methylpyridin-3-yl)ethyl]-3,4-dihydro-1 H-pyrido[4,3- b]indole, (hereinafter referred to as "latrepirdine") has the following structure:

The term "latrepirdine" should be understood, unless otherwise indicated herein, to include any pharmaceutically acceptable form of the compound. Latrepirdine may be present in crystalline or amorphous form.
The Amet metabolite of latrepirdine is 5-[2-(2,8-dimethyl-1 ,2,3,4-tetrahydro-5/-/- pyrido[4,3-ib]indol-5-yl)ethyl]pyridine-2-carboxylic acid, herein after referred to as "Amet", and has the following structure:

The Amet metabolite is a metabolite produced in humans when latrepirdine is orally dosed. Amet determination in human plasma samples is completed by solid phase extraction and analyzed by liquid chromatography/tandem mass spectrometry.
Latrepirdine and methods for synthesizing it are disclosed in PCT Publication
No. WO 2009/111540, filed March 4, 2008. Latrepirdine is useful in the treatment of various disorders, such as Alzheimer's disease, neurodegenerative disorders, Huntington's Disease and schizophrenia (see for example, U.S. Pat. No. 6,187,785; and U.S. Pat. Appl. Pub. Nos. 2007/0117835, 2007/0117834 and 2007/0225316).
In one embodiment of the invention a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but less than 0.1. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1.
In another embodiment, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed. For example, if 60 mg latrepirdine sustained release is given QD
in a single day, and the mean plasma latrepirdine AUC0-inf was determined to be 27 ng-hr/mL, this would equate to a mean AUC0-inf per mg of latrepirdine of 27 ng- hr/mL/60 mg latrepirdine = 0.45 ng-hr/mL per mg of latrepirdine.
In another embodiment of invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed. For example, if 60 mg latrepirdine sustained release is given QD in a single day, and the maximum plasma concentration (Cmax) was determined to be 6 ng/mL, this would equate to a mean maximum plasma concentration (Cmax) per mg of latrepirdine of the following: 6 ng/mL /60 mg latrepirdine = 0.1 ng/mL per mg of latrepirdine. In another embodiment, the Cmax is less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, the Cmax is less than about 0.25 ng/ml per mg of latrepirdine dosed.
In another embodiment of the invention a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a ratio of the mean area under the latrepirdine plasma concentration versus time curve (AUC0-inf) to the mean maximum latrepirdine plasma concentration (Cmax) of greater than about 10. For example, if 60 mg latrepirdine sustained release is given QD in a single day, and the mean plasma latrepirdine AUC0-inf was determined to be 80 ng-hr/mL and the maximum plasma concentration (Cmax) was determined to be 3.67 ng/mL, this would equate to a ratio of the mean area under the latrepirdine plasma concentration versus time curve (AUC0-inf) to the mean maximum latrepirdine plasma concentration (Cmax) of: AUC0-inf/Cmax = 80 ng-hr/mL/3.67 ng/mL = 21 .8.
In another embodiment, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, as a single dose in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-
inf) of between about 0.78 ng-hr/mL per mg of latrepirdine dosed and about 1.57 ng- hr/mL per mg of latrepirdine dosed and has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.12 ng/ml per mg of latrepirdine dosed. For example, if 60 mg latrepirdine sustained release is given QD in a single day, and the mean plasma latrepirdine AUC0-inf was determined to be 59.99 ng-hr/mL, and the maximum plasma concentration (Cmax) was determined to be 3.1 1 ng/mL, this would equate to a mean AUC0-inf per mg of latrepirdine of 59.99 ng-hr/mL/60 mg latrepirdine = 1 .0 ng-hr/mL per mg of latrepirdine and a mean maximum plasma concentration (Cmax) per mg of latrepirdine of the following: 3.1 1 ng/mL /60 mg latrepirdine = 0.05 ng/mL per mg of latrepirdine dosed.
In another embodiment of the invention a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean latrepirdine plasma concentration twelve (12) hours post dose (d2) of greater than about 0.040 ng/ml per mg of latrepirdine dosed. For example, if 60 mg latrepirdine sustained release is given QD in a single day, and the mean plasma concentration twelve hours post dose (C12) was determined to be 3.31 ng/mL, this would equate to a mean plasma concentration twelve hours post dose (C12) per mg of latrepirdine of the following: 3.31 ng/mL /60 mg latrepirdine = 0.055 ng/mL per mg of latrepirdine.
In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1 .4.
In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose relative bioavailability test/reference crossover study, using a 20 mg latrepirdine immediate release tablet as the reference control, displays a dose normalized relative
bioavailability greater than about 1 10%. The immediate release control tablet formulation is the Reference treatment and the sustained release formulations are the Test treatments.
In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours. In another embodiment of the invention, a sustained release pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form, when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours. In another embodiment of the invention, a sustained release pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form, when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours. In another embodiment of the invention, a sustained release pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form, when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours.
In one embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but less than 0.1 , and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1 . In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1. In another embodiment of the invention, a pharmaceutical dosage form
comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1 . In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1 . In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area
under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1.
In another embodiment, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a
CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours.
In another embodiment of invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of
latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed.
In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 8 hours and no
greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours.
In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage is a sustained release form, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage is a sustained release form, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage is a sustained release form, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours. In another embodiment of the invention, a sustained release pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form, when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours.
In one embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area
under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1 . In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1 . In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a
CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1.
In another embodiment, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of
latrepirdine dosed, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours.
In another embodiment of invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects,
having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed.
In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 6 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier,
wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 8 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, releases 80% of the latrepirdine in no less than 12 hours and no greater than 20 hours.
In another embodiment of the invention a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
In one embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the
plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1.
In another embodiment, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
In another embodiment of invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed.
In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the
latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
In one embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1 . In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1.
In another embodiment, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
In another embodiment of invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed.
In another embodiment of the invention, a pharmaceutical dosage form comprises and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 1 , displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that
80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 1 , displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 1 , displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours. In another embodiment of the invention, a sustained release pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form, when tested in Dissolution Test 1 , displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours. In another embodiment of the invention, a sustained release pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form, when tested in Dissolution Test 1 , displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours. In another embodiment of the invention, a sustained release pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form, when tested in Dissolution Test 1 , displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours.
In one embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a
CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1 . In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1 . In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , displays a lag phase
of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 1 , displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean
area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1.
In one embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUCo- inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUCo-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 2 hours in which less
than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no
greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC nf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours.
In one embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than
about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total
amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 1 , displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed.
In one embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , displays a lag phase
between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1 .4, and when tested in Dissolution Test 1 , displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form,
and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1 .4, and when tested in Dissolution Test 1 , displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4, and when tested in Dissolution Test 1 , displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours.
In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 2, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical
dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 2, displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 2, displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 2, displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in Dissolution Test 2, displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when tested in
Dissolution Test 2, displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours.
In one embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC0-inf) ratio of
latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1 . In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1 . In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form,
and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and when tested in Dissolution Test 2, displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1. In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUCo-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 , and
when tested in Dissolution Test 2, displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of Latrepirdine at a rate such that 80% of the total amount of Latrepirdine is released in no less than 7 hours and no greater than 20 hours. In another embodiment, the dosage form has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.01 but less than 0.1 . In a further embodiment, the dosage form has a the mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite greater than about 0.04 but less than 0.1 .
In one embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6
EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours. In another embodiment of the invention, a a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of
latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours.
In one embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when
tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted
state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of Latrepirdine is released in no less than 6 hours and no greater than 20 hours. In another embodiment, the dosage form has a Cmax less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet another embodiment, dosage form has a Cmax less than about 0.25 ng/ml per mg of latrepirdine dosed. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when tested in Dissolution Test 2, displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours. In other embodiments, the Cmax is less than about 0.50 ng/ml per mg of latrepirdine dosed. In yet other embodiments, the Cmax is less than about 0.25 ng/ml per mg of latrepirdine dosed.
In one embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a
sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, displays a lag phase of 2 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 3 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, displays a lag phase of 3 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a
rate such that 80% of the total amount of latrepirdine is released in no less than 4 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, displays a lag phase of 4 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 5 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1 .4, and when tested in Dissolution Test 2, displays a lag phase of 5 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 6 hours and no greater than 20 hours. In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUCo-inf) of 0.7 to 1.4, and when tested in Dissolution Test 2, displays a lag phase of 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 7 hours and no greater than 20 hours.
It is understood that any dosage form that does not meet the requirements of either Dissolution Test 1 or 2 as disclosed in the embodiments herein stated above is
not part of this invention. And any dosage form that does meet either Dissolution Test 1 or 2 is part of this invention.
In another embodiment of the invention a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and the total daily dose of latrepirdine needed for efficacy ranges from 1mg/day to 150 mg/day.
In another embodiment of the invention a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and the total daily dose of latrepirdine needed for efficacy ranges from 5mg/day to 100 mg/day.
In another embodiment of the invention a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and the total daily dose of latrepirdine needed for efficacy ranges from 10mg/day to 75 mg/day.
In another embodiment of the invention a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and the total daily dose of latrepirdine needed for efficacy ranges from 15 mg/day to 60 mg/day.
In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and said latrepirdine is embedded in a matrix which releases latrepirdine by diffusion. In another embodiment of this invention, a portion of the outside surface of said matrix is covered with an impermeable coating and the remainder of said outside surface is uncovered. In another embodiment, this dosage form is in the form of a tablet and the uncovered surface is in the form of an opening through said impermeable coating. In another embodiment, this dosage form is in the form of a tablet and the uncovered surface is in the form of a passageway which penetrates through the entire tablet. In another embodiment, this dosage form is in the form of a tablet and the uncovered surface is in the form of one or more slits through said impermeable coating or in the form of one or more strips removed therefrom. In another embodiment, this dosage form is substantially in the shape of a cylinder and said impermeable coating covers one or
both of the opposing flat surfaces thereof. This impermeable coating may alternatively only coat the radial surface thereof. In another embodiment, the matrix of this dosage form remains substantially intact during the period of latrepirdine release.
In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and said latrepirdine is embedded in a matrix which releases latrepirdine by eroding. In another embodiment, the matrix of this dosage form comprises hydroxypropyl methylcellulose. In another embodiment, the matrix of this dosage form comprises poly(ethylene oxide). In another embodiment, the matrix of this dosage form comprises polyacrylic acid.
In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and a reservoir of latrepirdine is encased in a membrane which limits the release rate of latrepirdine by diffusion. In one embodiment of the invention, this dosage form is in the form of a tablet coated with a membrane.
In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form and is in the form of a multiparticulate comprising particles, which particles are independently coated with a membrane which limits the release rate of latrepirdine by diffusion.
In another embodiment of the invention, a pharmaceutical dosage form comprises a core containing latrepirdine and a semi-permeable membrane coating wherein said dosage form is a sustained release dosage form, and said coating comprises a substantially water-insoluble polymer and a solid, water-soluble polymeric material. In a more specific embodiment of this invention, said dosage form delivers latrepirdine primarily by osmotic pressure. In another embodiment of this invention, the semi-permeable membrane is fabricated by an asymmetric membrane technology. In another embodiment of this invention, said water insoluble polymer comprises a cellulose derivative. One example of a cellulose derivative is cellulose acetate. In another embodiment of this invention, the solid water soluble polymeric
material comprises a polymer having an average molecular weight between 2000 and 50,000 daltons. The solid, water soluble polymeric material is selected from the group consisting of water soluble cellulose derivatives, acacia, dextrin, guar gum, maltodextrin, sodium alginate, starch, polyacrylates, and polyvinyl alcohols. Solid water soluble cellulose derivatives include but are not limited to hydroxypropylcellulose, hydroxypropylmethylcellulose and hydroxyethylcellulose. In another embodiment of the invention, the core of the sustained release pharmaceutical dosage form also contains a sugar. An example of such a sugar is mannitol.
In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine, a pharmaceutically acceptable carrier, and a CYP2D6 inhibitor, wherein said dosage form can be a sustained release dosage form or an immediate release dosage form. CYP2D6 inhibitors include but are not limited to: amiodarone, amitriptyline, bupropion, celecoxib, chlorpheniramine, chlorpromazine, cimetidine, cinacalcet, citalopram, chlorpheniramine, clomipramine, desipramine, diphenhydramine, doxepin, duloxetine, fluvoxamine, fluoxetine, goldenseal, halofantrine, haloperidol, hydroxyzine, imipramine, methadone, metoclopramide, moclobemide, paroxetine, pimozide, propafenone, quinidine/quinine, ritonavir, sertraline, terbinafine, thioridazine and ticlopidine, or pharmaceutically acceptable salts thereof. An additional CYP2D6 inhibitor includes St. John's wort, or an extract or constituent thereof. (See Summary Of Information On Human Cyp Enzymes: Human P450 Metabolism Data by Slobodan Rendic; Drug Metabolism 34(1 &2), 83-448 (2002).)
Some drugs and, in particular latrepirdine, are metabolized by cytochrome P450 (CYP) enzymes leading to unfavorable pharmacokinetics and the need for more frequent and higher doses than are typically desirable. Administration of such drugs with an agent that inhibits metabolism by CYP enzymes will improve the pharmacokinetics (e.g., increase half-life, increase the time to peak plasma concentration, increase blood levels) of the drug. It has been discovered that co- administration of latrepirdine with a drug or compound which is metabolized by CYP enzymes, especially the CYP 2D6 isozyme, causes an improvement in the pharmacokinetics of latrepirdine. In particular, it has been discovered that
coadministration of latrepirdine with a CYP2D6 inhibitor causes an unexpected improvement in the pharmacokinetics of latrepirdine. The ability of CYP2D6 inhibitors to improve the pharmacokinetics of latrepirdine can be demonstrated by the test method described herein.
In another embodiment the present invention provides methods of treating neurological and psychiatric disorders comprising: administering to a mammal a pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and the amount of latrepirdine is effective for treating such disorders. Neurological and psychiatric disorders, include but are not limited to: acute neurological and psychiatric disorders such as cerebral deficits subsequent to cardiac bypass surgery and grafting, stroke, cerebral ischemia, spinal cord trauma, head trauma, perinatal hypoxia, cardiac arrest, hypoglycemic neuronal damage, dementia, AIDS-induced dementia, vascular dementia, mixed dementias, age associated memory impairment, Alzheimer's disease, Huntington's Chorea, amyotrophic lateral sclerosis, ocular damage, retinopathy, cognitive disorders, including cognitive disorders associated with schizophrenia and bipolar disorders, idiopathic and drug- induced Parkinson's disease, muscular spasms and disorders associated with muscular spasticity including tremors, epilepsy, convulsions, migraine, migraine headache, urinary incontinence, substance tolerance, substance withdrawal, withdrawal from opiates, nicotine, tobacco products, alcohol, benzodiazepines, cocaine, sedatives, and hypnotics, psychosis, mild cognitive impairment, amnestic cognitive impairment, multi-domain cognitive impairment, obesity, schizophrenia, anxiety, generalized anxiety disorder, social anxiety disorder, panic disorder, post-traumatic stress disorder, obsessive compulsive disorder, mood disorders, depression, mania, bipolar disorders, trigeminal neuralgia, hearing loss, tinnitus, macular degeneration of the eye, emesis, brain edema, pain, acute and chronic pain states, severe pain, intractable pain, neuropathic pain, post-traumatic pain, tardive dyskinesia, sleep disorders, narcolepsy, attention deficit hyperactivity disorder, autism, Asperger's disease, and conduct disorder in a mammal. Accordingly, in one embodiment, the invention provides a method for treating a condition in a mammal, such as a human, selected from the conditions above, comprising administering a pharmaceutical
dosage form comprising latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form, and the amount of latrepirdine is effective for treating such disorders, to the mammal. The mammal is preferably a mammal in need of such treatment. As examples, the invention provides a method for treating Huntington's disease, attention deficit hyperactivity disorder, schizophrenia and Alzheimer's Disease.
In another embodiment, the present invention provides methods of treating neurological and psychiatric disorders comprising: administering to a patient in need thereof a pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is a sustained release dosage form and the amount of latrepirdine is effective for treating such disorders. In another embodiment the pharmaceutical dosage form is an immediate release dosage form and the sustained release or immediate release pharmaceutical dosage form is optionally used in combination with a CYP2D6 inhibitor to treat neurological and psychiatric disorders. The sustained release pharmaceutical dosage form is optionally used in combination with another active agent. Such an active agent may be, for example, an atypical antipsychotic, a cholinesterase inhibitor, or an NMDA receptor antagonist. Such atypical antipsychotics include, but are not limited to, ziprasidone, clozapine, olanzapine, risperidone, quetiapine, aripiprazole, paliperidone; such NMDA receptor antagonists include but are not limited to memantine; and such cholinesterase inhibitors include but are not limited to donepezil and galantamine.
In another embodiment the present invention provides a method for improving the pharmacokinetics of latrepirdine comprising administering to a human in need of such treatment a therapeutically effective amount of a combination of latrepirdine and a CYP2D6 inhibitor or a pharmaceutically acceptable salt thereof. In another embodiment the present invention provides a method of sustaining latrepirdine action in a human comprising orally administering to the human a therapeutically effective amount of a combination of latrepirdine and a CYP2D6 inhibitor or a
pharmaceutically acceptable salt thereof wherein the combination of latrepirdine and a CYP2D6 inhibitor results in a sustained action of latrepirdine as compared to administration of latrepirdine in the absence of a CYP2D6 inhibitor. In another
embodiment the present invention provides a method of treating a neurological and psychiatric disorder comprising administering to a human in need of such treatment a therapeutically effective amount of a combination of latrepirdine and a CYP2D6 inhibitor or a pharmaceutically acceptable salt thereof.
In another embodiment of the invention, a pharmaceutical dosage form comprises latrepirdine, a CYP2D6 inhibitor, and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than 0.5 but not greater than 15.
Unit dosage forms comprising latrepirdine and a CYP2D6 inhibitor are also embraced. Unit dosage forms contain a predetermined dose of latrepirdine and a predetermined dose of a CYP2D6 inhibitor. Preferably, a unit dosage form contains a predetermined dose of latrepirdine that is a therapeutically effective dose and a predetermined dose of a CYP2D6 inhibitor that reduces the rate of metabolism of latrepirdine as compared to the rate of metabolism of latrepirdine that would occur in the absence of a CYP2D6 inhibitor.
Kits comprising latrepirdine, a CYP2D6 inhibitor and instructions for use are also provided, where the kits may contain latrepirdine and a CYP2D6 inhibitor as a unit dosage form or a separately packaged components.
In another embodiment, a pharmaceutical dosage form comprises latrepirdine, a CYP2D6 inhibitor, and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of at least 10.2 ng-hr/mL per mg of latrepirdine dosed.
In another embodiment of invention, a pharmaceutical dosage form comprises latrepirdine, a CYP2D6 inhibitor, and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted
state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 3 ng/mL per mg of latrepirdine dosed.
In other embodiments of the invention the sustained release pharmaceutical dosage forms described above can comprise latrepirdine di-hydrochloride. In additional embodiments of the invention, the sustained release pharmaceutical dosage forms described above can be in the form of a tablet, capsule, multiparticulate, or beads. In other embodiments, any of the sustained release pharmaceutical dosage forms described can comprise a controlled release component; a delayed release and controlled release component; a delayed release and immediate release component or any combination thereof. In other embodiments of the invention, the sustained release pharmaceutical dosage forms can comprise one of the following delivery technologies or a combination thereof: matrix, osmotic, reservoir, delayed release, enteric coated, or pulsatile systems.
Sustained release dosage forms comprising latrepirdine and prepared by a method detailed herein are provided. Methods of preparing sustained release dosage forms comprising latrepirdine are also embraced.
DEFINITIONS
By "sustained release" is meant broadly that latrepirdine is released from an oral dosage form at a rate that is slower than immediate release. By "immediate release" is meant that at least 70% of the latrepirdine contained in the oral dosage form is released within 1 hour when tested in Dissolution Test 1. It is understood that oral dosage forms containing latrepirdine as the only active agent in an immediate release -only dosage form are not part of this invention. Oral dosage form is intended to embrace tablets, capsules, multiparticulates or beads. "Sustained release" is intended to embrace an oral composition that consists of either one or a combination of the following:
a) a controlled release component alone;
b) a delayed release and controlled release component;
c) a delayed release and immediate release component
The term "latrepirdine" is interchangeable with "2,8-dimethyl-5-[2-(6- methylpyridin-3-yl)ethyl]-3,4-dihydro-1 /-/-pyrido[4,3-b]indole" and should be
understood herein, unless otherwise indicated herein, to include any pharmaceutically acceptable form of the compound.
By "pharmaceutically acceptable form" is meant any pharmaceutically acceptable form, including, solvates, hydrates, isomorphs, polymorphs, co-crystals, pseudomorphs, neutral forms, acid addition salt forms, and prodrugs. The pharmaceutically acceptable acid addition salts of latrepirdine are prepared in a conventional manner by treating a solution or suspension of the free base with about one or two chemical equivalents of a pharmaceutically acceptable acid. Conventional concentration and recrystallization techniques are employed in isolating the salts. Illustrative of suitable acids are acetic, lactic, succinic, maleic, tartaric, citric, gluconic, ascorbic, mesylic, tosylic, benzoic, cinnamic, fumaric, sulfuric, phosphoric, hydrochloric, hydrobromic, hydroiodic, sulfamic, sulfonic such as methanesulfonic, benzenesulfonic, and related acids. Some preferred forms of latrepirdine include the free base and latrepirdine dihydrochloride dihydrate.
The "solid oral dosage form" of the present invention is a pharmaceutically- acceptable solid oral dosage form, meaning that the dosage form is safe for administration to humans and all excipients in the dosage form are pharmaceutically- acceptable, in other words safe for human ingestion.
By "administered orally to a cohort of subjects having a CYP2D6 EM status in the fasted state" is meant administration of latrepirdine either once (QD) or twice (BID - dosed about 12 hours apart) over a 24 hour period. Prior to the initial administration of latrepirdine, the subject has not been administered latrepirdine for a sufficient period of time (minimally 7 days) so that the subject's plasma concentration of latrepirdine is below the detectable limit.
"Cohort" refers to a standard testing population. Such testing populations are well known in the art and are designed to produce meaningful reproducible results. One common size is about 20 healthy subjects, equally divided between 10 males and 10 females.
"CYP2D6 EM status" refers to subjects, preferably human, who have been determined by genotyping to be CYP2D6 Extensive metabolizers. They represent approximately 60-80% of the population and therefore enrolling sufficient EM subjects to run a single dose pharmacokinetic study is easily achievable. The
importance of genotyping subjects is due to the variability that can be observed between poor metabolizers (PMs) and ultra metabolizers (UMs) for compounds which are metabolized extensively by CYP2D6. To identify Poor Metabolizers (PMs), Intermediate Metabolizers (IMs), Extensive Metabolizers (EMs), and Ultra Rapid Metabolizers (UMs), easy, reliable tests which require only a blood sample and less than a day for analysis are now available. One such example, not meant to be inclusive of all tests available, is Roche's AmpliChip CYP450 Test which was approved by the FDA in 2005. The AmpliChip test provides a predicted phenotype of the subject in one of the following four categories; Poor Metabolizers (PMs), Intermediate Metabolizers (IMs), Extensive Metabolizers (EMs) and Ultra Rapid Metabolizers.
A study to measure the concentration of latrepirdine in the plasma after initial administration may be conducted using conventional methods for making such a determination. The study should include at least 20 subjects, preferably human, which have all been genotyped to ensure that they have CYP2D6 EM status, in order to measure mean values for Cmax and AUC0-inf- The study should be conducted in the fasted state. Prior to the initial administration of latrepirdine, the subject has not been administered latrepirdine for a sufficient length of time (minimally 7 days) so that the subject's plasma concentration of latrepirdine prior to administration of the dosage form is below the detectable limit. Plasma samples are taken at a sufficient number of time points to determine Cmax and AUC0-inf-
The phrase "fasted" as used herein is defined as follows: the dosing state which is defined following an overnight fast (wherein 0 caloric intake has occurred) of at least 10 hours. Subjects are administered the dosage form with 240 imL of water. No food should be allowed for at least 4 hours post-dose. Water can be allowed as desired except for one hour before and after drug administration.
By "mean area under the plasma concentration versus time curve ratio of latrepirdine to its Amet metabolite" is meant, the individual ratio of the mean area under the plasma concentration versus time curve of latrepirdine (e.g. AUC0-inf) to the mean area under the plasma concentration versus time curve of its Amet metabolite (e.g. AUCo-inf) is first calculated for each subject, and then the corresponding individual ratios are averaged together. In this way, it is the average of each
corresponding individuals ratio which is determined. The latrepirdine and Amet concentrations are measured by standard liquid chromatography / tandem mass spectroscopy techniques.
The phrase "fed" as used herein is defined as follows: the dosing state which is defined following an overnight fast (wherein 0 caloric intake has occurred) of at least 10 hours, subjects then begin the recommended high fat meal 30 minutes prior to administration of the drug product. Subjects should eat this meal in 30 minutes or less; however the drug product should be administered 30 minutes after the start of the meal. The drug product should be administered with 240 imL of water. No food should be allowed for at least 4 hours post-dose. Water can be allowed as desired except for one hour before and after drug administration. A high fat (approximately 50 percent of the total caloric content of the meal is derived from fat) and high calorie (approximately 800 to 1000 calories) meal should be used as the test meal under the fed condition. This test meal should derive approximately 150, 250, and 500-600 calories from protein, carbohydrate, and fat respectively. An example test meal would be two eggs fried in butter, two strips of bacon, two slices of toast with butter, four ounces of hash brown potatoes and eight ounces of whole milk.
In a single dose fed / fasted crossover clinical study, the test latrepirdine solid oral dosage form can be administered to a cohort of subjects who have not eaten any food for at least ten hours and will not eat any food for at least four hours after administration of the dosage form. For comparison's sake, another cohort of subjects can be administered an identical latrepirdine solid oral dosage form in a fed state, for example about 30 minutes after they begin eating a United States Food and Drug Administration (FDA) standard high fat breakfast, or other meal containing a quantity of fat and calories comparable thereto, the cohort completing eating the breakfast or other meal within about 30 minutes or less of beginning the breakfast or other meal. The cohorts can subsequently be "switched", with the cohort which had been tested with the fasted protocol being tested according to the fed protocol, and the cohort which had been tested with the fed protocol being tested according to the fasted protocol. It is recommended that a period of time, sometimes referred to as a "washout period", of greater than four days from completion of the first dosing pass before switching the fed or fasted protocol for each cohort.
The calculation of the mean area under the serum concentration versus time curve (AUC) is a well-known procedure in the pharmaceutical arts and is described, for example, in Welling, "Pharmacokinetics Processes and Mathematics," ACS Monograph 185 (1986). To determine the mean fed/fasted ratio, the individual ratio of the mean area under the plasma concentration versus time curve of latrepirdine (e.g. AUCo-inf) in the fed state to the mean area under the plasma concentration versus time curve of latrepirdine (e.g. AUC0-inf) in the fasted state is first calculated, and then the corresponding individual ratios are averaged together. In this way, it is the average of each corresponding individual's ratio which is determined.
"Dissolution Test 1 " refers to the following test of dosage forms of latrepirdine.
The dissolution test is conducted in a standard USP rotating paddle apparatus as disclosed in United States Pharmacopoeia (USP) Dissolution Test Chapter 711 , Apparatus 2. Paddles are rotated at 50 rpm and the dosage form is added to 500 mL of 0.2M pH 6.8 potassium phosphate buffer at 37° C. At appropriate times following test initiation (e.g., insertion of the dosage form into the apparatus), filtered aliquots (typically 1.5 mL) from the test medium are analyzed for latrepirdine by high performance liquid chromatography (HPLC). Dissolution results are reported as the percent of the total dose of latrepirdine tested dissolved versus time.
"Dissolution Test 2" refers to the following test of dosage forms of latrepirdine. The test is modified from that detailed above as follows: Paddles are rotated at 100 rpm and dissolution is conducted in two stages at 37° C. Dosage forms are added to a first test medium comprising 500 mL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 mL of a dibasic and monobasic sodium phosphate buffer (total phosphate strength is 0.425M, pH 7.6), thereby converting the acid media from the first stage to a buffer having a pH of 7.5. At appropriate times following test initiation (e.g., insertion of the dosage form into the apparatus), filtered aliquots (typically 1.5 mL) from the test medium are analyzed for latrepirdine by high performance liquid chromatography (HPLC). Dissolution results are reported as the percent of the total dose of latrepirdine tested dissolved versus time.
By "relative bioavailability test/reference crossover study" is meant, the individual ratio of the dose normalized mean area under the plasma concentration
versus time curve of the latrepirdine sustained release test formulation (e.g. AUC0-inf) to the dose normalized mean area under the plasma concentration versus time curve of the latrepirdine immediate release reference formulation (e.g. AUC0-inf) is first calculated, and then the corresponding individual ratios are averaged together. In this way, it is the average of each corresponding individual's ratio which is determined. The relative bioavailability ratio of the latrepirdine Test/Reference is expressed as a simple ratio of the dose normalized mean area under the plasma concentration versus time curve of the latrepirdine test formulation (e.g. sustained release formulation) (AUC0-inf ng-hr/mL per mg latrepirdine)/the dose normalized mean area under the plasma concentration versus time curve of the latrepirdine reference formulation (immediate release formulation) (AUC0-inf ng-hr/mL per mg latrepirdine).
Sustained Release - Matrix Systems
In one embodiment, latrepirdine is incorporated into an erodible or non- erodible polymeric matrix tablet. By an erodible matrix is meant aqueous-erodible or water-swellable or aqueous-soluble in the sense of being either erodible or swellable or dissolvable in pure water or requiring the presence of an acid or base to ionize the polymeric matrix sufficiently to cause erosion or dissolution. When contacted with the aqueous use environment, the erodible polymeric matrix imbibes water and forms an aqueous-swollen gel or "matrix" that entraps the latrepirdine. The aqueous- swollen matrix gradually erodes, swells, disintegrates, disperses or dissolves in the environment of use, thereby controlling the release of latrepirdine to the environment of use. Examples of such dosage forms are well known in the art. See, for example, Remington: The Science and Practice of Pharmacy, 20th Edition, 2000.
A key ingredient of the water-swollen matrix is the water-swellable, erodible, or soluble polymer, which may generally be described as an osmopolymer, hydrogel or water-swellable polymer. Such polymers may be linear, branched, or crosslinked. They may be homopolymers or copolymers. Exemplary polymers include naturally occurring polysaccharides such as chitin, chitosan, dextran and pullulan; gum agar, gum arabic, gum karaya, locust bean gum, gum tragacanth, carrageenans, gum ghatti, guar gum, xanthan gum and scleroglucan; starches such as dextrin and maltodextrin; hydrophilic colloids such as pectin; alginates such as ammonium
alginate, sodium, potassium or calcium alginate, propylene glycol alginate; gelatin; collagen; and cellulosics. By "cellulosics" is meant a cellulose polymer that has been modified by reaction of at least a portion of the hydroxyl groups on the saccharide repeat units with a compound to form an ester-linked or an ether-linked substituent. For example, the cellulosic ethyl cellulose has an ether linked ethyl substituent attached to the saccharide repeat unit, while the cellulosic cellulose acetate has an ester linked acetate substituent.
Cellulosics for the erodible matrix comprise aqueous-soluble and aqueous- erodible cellulosics such as ethyl cellulose (EC), methylethyl cellulose (MEC), carboxymethyl cellulose (CMC), carboxymethyl ethylcellulose (CMEC), hydroxyethyl cellulose (HEC), hydroxypropyl cellulose (HPC), cellulose acetate phthalate (CAP), cellulose acetate trimellitate (CAT), hydroxypropyl methyl cellulose (HPMC), hydroxypropyl methyl cellulose phthalate (HPMCP), hydroxypropyl methyl cellulose acetate succinate (HPMCAS), hydroxypropyl methyl cellulose acetate trimellitate (HPMCAT), and ethylhydroxy ethylcellulose (EHEC).
A particularly preferred class of such cellulosics comprises various grades of low viscosity (MW less than or equal to 50,000 daltons) and high viscosity (MW greater than 50,000 daltons) HPMC. Commercially available low viscosity HPMC polymers include the Dow METHOCEL™ series E3, E5, E15LV, E50LV and K100LV, while high viscosity HPMC polymers include E4MCR, E10MCR, K4M, K15M and K100M; especially preferred in this group are the METHOCEL™ K series. Other commercially available types of HPMC include the Shin Etsu METOLOSE™ 90SH series. In one embodiment, the HPMC has a low viscosity, meaning that the viscosity of a 2% (w/v) solution of the HPMC in water is less than about 120 cp. A preferred HPMC is one in which the viscosity of a 2% (w/v) solution of the HPMC in water ranges from 80 to 120 cp (such as METHOCEL™ K100LV).
Other materials useful as the erodible matrix material include, but are not limited to, pullulan, polyvinyl pyrrolidone, polyvinyl alcohol, polyvinyl acetate, glycerol fatty acid esters, polyacrylamide, polyacrylic acid, copolymers of ethacrylic acid or methacrylic acid (EUDRAGIT®, Rohm America, Inc., Piscataway, New Jersey) and other acrylic acid derivatives such as homopolymers and copolymers of
butylmethacrylate, methylmethacrylate, ethylmethacrylate, ethylacrylate, (2-dimethylaminoethyl)methacrylate, and (trimethylaminoethyl) methacrylate chloride.
The erodible matrix polymer may also contain additives and excipients known in the pharmaceutical arts, including osmopolymers, osmagens, solubility-enhancing or -retarding agents and excipients that promote stability or processing of the dosage form.
Alternatively, the controlled-release portion may comprise a non-erodible matrix. In such dosage forms, latrepirdine is distributed in an inert matrix. The drug is released by diffusion through the inert matrix. Examples of materials suitable for the inert matrix include insoluble plastics, such as copolymers of ethylene and vinyl acetate, methyl acrylate-methyl methacrylate copolymers, polyvinyl chloride, and polyethylene; hydrophilic polymers, such as ethyl cellulose, cellulose acetate, and crosslinked polyvinylpyrrolidone (also known as crospovidone); and fatty compounds, such as carnauba wax, microcrystalline wax, and triglycerides. Such dosage forms are described further in Remington: The Science and Practice of Pharmacy, 20th edition (2000).
In another embodiment, a matrix multiparticulate, comprises a plurality of latrepirdine-containing particles, each particle comprising a mixture of latrepirdine with one or more excipients selected to form a matrix capable of limiting the dissolution rate of the latrepirdine into an aqueous medium. The matrix materials useful for this embodiment are generally water-insoluble materials such as waxes, cellulose, or other water-insoluble polymers. If needed, the matrix materials may optionally be formulated with water-soluble materials which can be used as binders or as permeability-modifying agents. Matrix materials useful for the manufacture of these dosage forms include microcrystalline cellulose such as Avicel (registered trademark of FMC Corp., Philadelphia, Pa.), including grades of microcrystalline cellulose to which binders such as hydroxypropyl methyl cellulose have been added, waxes such as paraffin, modified vegetable oils, carnauba wax, hydrogenated castor oil, beeswax, and the like, as well as synthetic polymers such as polyvinyl chloride), polyvinyl acetate), copolymers of vinyl acetate and ethylene, polystyrene, and the like. Water soluble binders or release modifying agents which can optionally be formulated into the matrix include water-soluble polymers such as hydroxypropyl
cellulose (HPC), hydroxypropyl methyl cellulose (HPMC), methyl cellulose, poly (N- vinyl-2-pyrrolidinone) (PVP), poly(ethylene oxide) (PEO), poly(vinyl alcohol) (PVA), xanthan gum, carrageenan, and other such natural and synthetic materials. In addition, materials which function as release-modifying agents include water-soluble materials such as sugars or salts. Preferred water-soluble materials include lactose, sucrose, glucose, and mannitol, as well as HPC, HPMC, and PVP.
A process for manufacturing matrix multiparticulates is the extrusion/spheronization process. For this process, the latrepirdine is wet-massed with a binder, extruded through a perforated plate or die, and placed on a rotating disk. The extrudate ideally breaks into pieces which are rounded into spheres, spheroids, or rounded rods on the rotating plate. Another process and composition for this method involves using water to wet-mass a blend comprising about 20 to 75% of micro-crystalline cellulose blended with, correspondingly, about 80 to 25% latrepirdine.
Another process for manufacturing matrix multiparticulates is the preparation of wax granules. In this process, a desired amount of latrepirdine is stirred with liquid wax to form a homogeneous mixture, cooled and then forced through a screen to form granules. Preferred matrix materials are waxy substances. Some preferred waxy substances are hydrogenated castor oil and carnauba wax and stearyl alcohol.
A further process for manufacturing matrix multiparticulates involves using an organic solvent to aid mixing of the latrepirdine with the matrix material. This technique can be used when it is desired to utilize a matrix material with an unsuitably high melting point that, if the material were employed in a molten state, would cause decomposition of the drug or of the matrix material, or would result in an unacceptable melt viscosity, thereby preventing mixing of latrepirdine with the matrix material. Latrepirdine and matrix material may be combined with a modest amount of solvent to form a paste, and then forced through a screen to form granules from which the solvent is then removed. Alternatively, latrepirdine and matrix material may be combined with enough solvent to completely dissolve the matrix material and the resulting solution (which may contain solid drug particles) spray dried to form the particulate dosage form. This technique is preferred when the matrix material is a high molecular weight synthetic polymer such as a cellulose ether or cellulose ester.
Solvents typically employed for the process include acetone, ethanol, isopropanol, ethyl acetate, and mixtures of two or more.
In one embodiment, the matrix multiparticulates are formed by the melt spray congeal process. The melt-congeal core comprises a matrix material. The matrix material serves two functions. First, the matrix material allows formation of relatively smooth, round cores that are amenable to coating. Second, the matrix material binds the optional excipients and/or drugs that may be incorporated into the core. The matrix material has the following physical properties: a sufficiently low viscosity in the molten state to form multiparticulates, as detailed below; and rapidly congeals to a solid when cooled below its melting point. For those multiparticulates incorporating drug in the core, the matrix preferably has a melting point below that of the melting point or decomposition point of the drug, and does not substantially dissolve the drug.
The melt-congeal cores consist essentially of a continuous phase of matrix material and optionally other excipients, with optional drug particles and optional swelling agent particles encapsulated within. Because of this, a sufficient amount of matrix material must be present to form smooth cores that are large enough to coat. In the case of cores containing solid particles, such as drug or swelling agent, the core must contain a sufficient amount of matrix material to encapsulate the drug and swelling agent to form relatively smooth and spherical cores, which are more easily coated by conventional spray-coating processes than irregularly-shaped ones. The matrix material may be present in the core from at least about 30 wt percent, at least about 50 wt percent, at least about 70wt percent, at least about 80 wt percent, at least about 90wt percent, and up to 100 wt percent based on the mass of the uncoated core.
In order to form small, smooth round cores, the matrix material must be capable of being melted and then atomized. The matrix material or mixture of materials is solid at 25 degrees C. However, the matrix material melts, or is capable of melting with the addition of an optional processing aid, at a temperature of less than 200 degrees centigrade so as to be suitable for melt-congeal processing described below. Preferably, the matrix material has a melting point between 50 degrees C and 150CC. Although the term "melt" generally refers to the transition of a
crystalline material from its crystalline to its liquid state, which occurs at its melting point, and the term "molten" generally refers to such a crystalline material in its fluid state, as used herein, the terms are used more broadly. In the case of "melt," the term is used to refer to the heating of any material or mixture of materials sufficiently that it becomes fluid in the sense that it may be pumped or atomized in a manner similar to a crystalline material in the fluid state. Likewise "molten" refers to any material or mixture of materials that is in such a fluid state.
The matrix material is selected from the group consisting of waxes, long chain alcohols (Ci2 or greater), fatty acid esters, glycolized fatty acid esters, phosphoglycerides, polyoxyethylene alkyl ethers, long chain carboxylic acids (Ci2 or greater), sugar alcohols, and mixtures thereof. Exemplary matrix materials include highly purified forms of waxes, such as Camauba wax, white and yellow beeswax, ceresin wax, microcrystalline wax, and paraffin wax; long-chain alcohols, such as stearyl alcohol, cetyl alcohol and polyethylene glycol; fatty acid esters (also known as fats or glycerides), such as isopropyl palmitate, isopropyl myristate, glyceryl monooleate, glyceryl monostearate, glyceryl palmitostearate, mixtures of mono-, di-, and trialkyl glycerides, including mixtures of glyceryl mono-, di-, and tribehenate, glyceryl tristearate, glyceryl tripalmitate and hydrogenated vegetable oils, including hydrogenated cottonseed oil; glycolized fatty acid esters, such as polyethylene glycol stearate and polyethylene glycol distearate; polyoxyethylene alkyl ethers; polyethoxylated castor oil derivatives; long-chain carboxylic acids such as stearic acid; and sugar alcohols such as mannitol and erythritol. The matrix material may comprise mixtures of materials, such as mixtures of any of the foregoing.
The core may also contain a variety of other excipients, present in the core in an amount of from 0 to 40 wt percent, based upon the mass of the uncoated core.
One preferred excipient is a dissolution enhancer, which may be used to increase the rate of water uptake by the core and consequent expansion of the swelling agent. The dissolution enhancer is a different material than the matrix material. The dissolution enhancer may be in a separate phase or a single phase with the matrix material. Preferably, at least a portion of the dissolution enhancer is phase-separated from the matrix material. As water enters the core, the dissolution-enhancer dissolves, leaving channels which allow water to more rapidly enter the core. In
general, dissolution enhancers are amphiphilic compounds and are generally more hydrophilic than the matrix materials. Examples of dissolution enhancers include: surfactants such as poloxamers, docusate salts, polyoxyethylene castor oil derivatives, polysorbates, sodium lauryl sulfate, and sorbitan monoesters; sugars, such as glucose, xylitol, sorbitol and maltitol; salts, such as sodium chloride, potassium chloride, lithium chloride, calcium chloride, magnesium chloride, sodium sulfate, potassium sulfate, sodium carbonate, magnesium sulfate and potassium phosphate; and amino acids, such as alanine and glycine; and mixtures thereof. One surfactant-type dissolution-enhancer is a poloxambetar (commercially available as the LUTROL or PLURONIC series from BASF Corp.).
The core may also contain other optional excipients, such as agents that inhibit or delay the release of drug from the multiparticulates. Such dissolution- inhibiting agents are generally hydrophobic and include dialkylphthalates such as dibutyl phthalate, and hydrocarbon waxes, such as microcrystalline wax and paraffin wax. Another useful class of excipients comprises materials that may be used to adjust the viscosity of the molten feed used to form the cores. Such viscosity- adjusting excipients will generally make up 0 to 25 wt percent of the core. The viscosity of the molten feed is a key variable in obtaining cores with a narrow particle size distribution. For example, when a spinning-disk atomizer is employed, it is preferred that the viscosity of the molten mixture be at least about 1 cp and less than about 10,000 cp, preferably at least 50 cp and less than about 1000 cp. If the molten mixture has a viscosity outside these ranges, a viscosity-adjusting agent can be added to obtain a molten mixture within the viscosity range. Examples of viscosity- reducing excipients include stearyl alcohol, cetyl alcohol, low molecular weight polyethylene glycol (i.e., less than about 1000 daltons), isopropyl alcohol, and water. Examples of viscosity-increasing excipients include microcrystalline wax, paraffin wax, synthetic wax, high molecular weight polyethylene glycols (i.e., greater than about 5000 daltons), ethyl cellulose, hydroxypropyl cellulose, hydroxypropyl methyl cellulose, methyl cellulose, silicon dioxide, microcrystalline cellulose, magnesium silicate, sugars, and salts.
For those embodiments containing a drug in the core, other excipients may be added to adjust the release characteristics of the drug from the cores. For
example, an acid or base may be included in the composition to modify the rate at which drug is released in an aqueous use environment. Examples of acids or bases that can be included in the composition include citric acid, adipic acid, malic acid, fumaric acid, succinic acid, tartaric acid, di- and tribasic sodium phosphate, di- and tribasic calcium phosphate, mono-, di-, and triethanolamine, sodium bicarbonate and sodium citrate dihydrate. Such excipients may make up 0 to 25 wt percent of the core, based on the total mass of the core.
Still other excipients may be added to improve processing, such as excipients to reduce the static charge on the cores or to reduce the melting temperature of the matrix material. Examples of such anti-static agents include talc and silicon dioxide. Flavorants, colorants, and other excipients may also be added in their usual amounts for their usual purposes. Such excipients may make up 0 to 25 wt percent of the core, based on the total mass of the core.
The multiparticulates are made via a melt-congeal process comprising the steps: (a) forming a molten mixture comprising the drug, the glyceride and the poloxamer; (b) delivering the molten mixture of step (a) to an atomizing means to form droplets from the molten mixture; and (c) congealing the droplets from step (b) to form multiparticulates.
The processing conditions are chosen to maintain the crystallinity of the drug. The temperature of the molten mixture is kept below the melting point of the drug. Preferably, at least 70 wt percent of the drug remains crystalline within the molten feed, more preferably, at least 80 wt percent and most preferably at least 90 wt percent.
The term "molten mixture" as used herein refers to a mixture of drug, glyceride, and poloxamer heated sufficiently that the mixture becomes sufficiently fluid that the mixture may be formed into droplets or atomized. Atomization of the molten mixture may be carried out using any of the atomization methods described below. Generally, the mixture is molten in the sense that it will flow when subjected to one or more forces such as pressure, shear, and centrifugal force, such as that exerted by a centrifugal or spinning-disk atomizer. Thus, the drug/glyceride/poloxamer mixture may be considered "molten" when any portion of the drug/glyceride/poloxamer mixture becomes sufficiently fluid that the mixture, as a
whole, may be atomized. Generally, a mixture is sufficiently fluid for atomization when the viscosity of the molten mixture is less than about 20,000 cp. Often, the mixture becomes molten when the mixture is heated above the melting point of the glyceride/poloxamer mixture, in cases where the glyceride/poloxamer mixture is sufficiently crystalline to have a relatively sharp melting point; or, when the glyceride/poloxamer mixture is amorphous, above the softening point of the glyceride/poloxamer mixture. The molten mixture is therefore often a suspension of solid particles in a fluid matrix. In one preferred embodiment, the molten mixture comprises a mixture of substantially crystalline drug particles suspended in a glyceride/poloxamer mixture that is substantially fluid. In such cases, a portion of the drug may be dissolved in the glyceride/poloxamer mixture and a portion of the glyceride/poloxamer mixture may remain solid.
Virtually any process may be used to form the molten mixture. One method involves heating the glyceride/poloxamer mixture in a tank until it is fluid and then adding the drug to the molten glyceride/poloxamer mixture. Generally, the glyceride/poloxamer mixture is heated to a temperature of about 10 degrees C. or more above the temperature at which it becomes fluid. When one or more of the glyceride/poloxamer components is crystalline, this is generally about 10 degrees C. or more above the melting point of the lowest melting point material of the mixture. The process is carried out so that at least a portion of the feed remains fluid until atomized. Once the glyceride/poloxamer mixture has become fluid, the drug may be added to the fluid carrier or "melt." Although the term "melt" generally refers specifically to the transition of a crystalline material from its crystalline to its liquid state, which occurs at its melting point, and the term "molten" generally refers to such a crystalline material in its fluid state, as used herein, the terms are used more broadly, referring in the case of "melt" to the heating of any material or mixture of materials sufficiently that it becomes fluid in the sense that it may be pumped or atomized in a manner similar to a crystalline material in the fluid state. Likewise "molten" refers to any material or mixture of materials that is in such a fluid state. Alternatively, the drug, the glyceride, and the poloxamer may be added to the tank and the mixture heated until the mixture has become fluid.
Once the glyceride/poloxamer mixture has become fluid and the drug has been added, the molten mixture is mixed to ensure the drug is uniformly distributed therein. Mixing is generally done using mechanical means, such as overhead mixers, magnetically driven mixers and stir bars, planetary mixers, and homogenizers. Optionally, the contents of the tank can be pumped out of the tank and through an inline, static mixer or extruder and then returned to the tank. The amount of shear used to mix the molten feed should be sufficiently high to ensure uniform distribution of the drug in the molten carrier. The amount of shear is kept low enough so the form of the drug does not change, i.e., so as to cause an increase in the amount of amorphous drug or a change in the crystalline form of the drug. It is also preferred that the shear not be so high as to reduce the particle size of the drug crystals. The molten mixture can be mixed from a few minutes to several hours, the mixing time being dependent on the viscosity of the feed and the solubility of drug and any optional excipients in the carrier.
An alternative method of preparing the molten mixture is to use two tanks, melting either the glyceride or the poloxamer in one tank and the other component in another tank. The drug is added to one of these tanks and mixed as described above. The two melts are then pumped through an in-line static mixer or extruder to produce a single molten mixture that is directed to the atomization process described below.
Another method that can be used to prepare the molten mixture is to use a continuously stirred tank system. In this system, the drug, glyceride, and poloxamer are continuously added to a heated tank equipped with means for continuous stirring, while the molten feed is continuously removed from the tank. The contents of the tank are heated such that the temperature of the contents is about 10 degrees C. or more above the melting point of the carrier. The drug, glyceride, and poloxamer are added in such proportions that the molten mixture removed from the tank has the desired composition. The drug is typically added in solid form and may be pre-heated prior to addition to the tank. The glyceride and poloxamer may also be preheated or even pre-melted prior to addition to the continuously stirred tank system.
In another method for forming the molten mixture is by an extruder. By "extruder" is meant a device or collection of devices that creates a molten extrudate
by heat and/or shear forces and/or produces a uniformly mixed extrudate from a solid and/or liquid (e.g., molten) feed. Such devices include, but are not limited to single- screw extruders; twin-screw extruders, including co-rotating, counter-rotating, intermeshing, and non-intermeshing extruders; multiple screw extruders; ram extruders, consisting of a heated cylinder and a piston for extruding the molten feed; gear-pump extruders, consisting of a heated gear pump, generally counter-rotating, that simultaneously heats and pumps the molten feed; and conveyer extruders. Conveyer extruders comprise a conveyer means for transporting solid and/or powdered feeds, such, such as a screw conveyer or pneumatic conveyer, and a pump. At least a portion of the conveyer means is heated to a sufficiently high temperature to produce the molten mixture. The molten mixture may optionally be directed to an accumulation tank, before being directed to a pump, which directs the molten mixture to an atomizer. Optionally, an in-line mixer may be used before or after the pump to ensure the molten mixture is substantially homogeneous. In each of these extruders the molten mixture is mixed to form a uniformly mixed extrudate. Such mixing may be accomplished by various mechanical and processing means, including mixing elements, kneading elements, and shear mixing by backflow. Thus, in such devices, the composition is fed to the extruder, which produces a molten mixture that can be directed to the atomizer.
In one embodiment, the composition is fed to the extruder in the form of a solid powder. The powdered feed can be prepared using methods well known in the art for obtaining powdered mixtures with high content uniformity. Generally, it is desirable that the particle sizes of the drug, glyceride, and poloxamer be similar to obtain a substantially uniform blend. However, this is not essential to the successful practice of the invention.
An example of a process for preparing a substantially uniform blend is as follows. First, the glyceride and poloxamer are milled so that their particle sizes are about the same as that of the drug; next, the drug, glyceride, and poloxamer are blended in a V-blender for 20 minutes; the resulting blend is then de-lumped to remove large particles; the resulting blend is finally blended for an additional 4 minutes. In some cases it is difficult to mill the glyceride and poloxamer to the desired particle size since many of these materials tend to be waxy substances and the heat
generated during the milling process can gum up the milling equipment. In such cases, small particles of the glyceride and poloxamer can be formed using a melt- or spray-congeal process, as described below. The resulting congealed particles of glyceride and poloxamer can then be blended with the drug to produce the feed for the extruder.
Another method for producing the feed to the extruder is to melt the glyceride and poloxamer in a tank, mix in the drug as described above for the tank system, and then cool the molten mixture, producing a solidified mixture of drug and carrier. This solidified mixture can then be milled to a uniform particle size and fed to the extruder. A two-feed extruder system can also be used to produce the molten mixture. In this system the drug, glyceride, and poloxamer, all in powdered form, are fed to the extruder through the same or different feed ports. In this way, the need for blending the components is eliminated.
Alternatively, the glyceride and poloxamer in powder form may be fed to the extruder at one point, allowing the extruder to melt the glyceride and poloxamer. The drug is then added to the molten glyceride and poloxamer through a second feed delivery port part way along the length of the extruder, thus minimizing the contact time of the drug with the molten glyceride and poloxamer. The closer the second feed delivery port is to the extruder exit, the lower is the residence time of drug in the extruder. Multiple-feed extruders can be used when optional excipients are included in the multiparticulate.
In another method, the composition is in the form of large solid particles or a solid mass, rather than a powder, when fed to the extruder. For example, a solidified mixture can be prepared as described above and then molded to fit into the cylinder of a ram extruder and used directly without milling.
In another method, the glyceride and poloxamer can be first melted in, for example, a tank, and fed to the extruder in molten form. The drug, typically in powdered form, may then be introduced to the extruder through the same or a different delivery port used to feed the glyceride and poloxamer into the extruder. This system has the advantage of separating the melting step for the glyceride and poloxamer from the mixing step, minimizing contact of the drug with the molten glyceride and poloxamer.
In each of the above methods, the extruder should be designed such that it produces a molten mixture with the drug crystals uniformly distributed in the glyceride/poloxamer mixture. Generally, the temperature of the extrudate should be about 10 degrees C. or more above the temperature at which the drug and carrier mixture becomes fluid. The various zones in the extruder should be heated to appropriate temperatures to obtain the desired extrudate temperature as well as the desired degree of mixing or shear, using procedures well known in the art. As discussed above for mechanical mixing, a minimum shear should be used to produce a uniform molten mixture, such that the crystalline form of the drug is unchanged and that dissolution or formation of amorphous drug is minimized.
The feed is preferably molten prior to congealing for at least 5 seconds, more preferably at least 10 seconds, and most preferably at least 15 seconds, so as to ensure adequate homogeneity of the drug/glyceride/poloxamer melt. It is also preferred that the molten mixture remain molten for no more than about 20 minutes to limit exposure of the drug to the molten mixture. As described above, depending on the reactivity of the chosen glyceride/poloxamer mixture, it may be preferable to further reduce the time that the mixture is molten to well below 20 minutes in order to limit drug degradation to an acceptable level. In such cases, such mixtures may be maintained in the molten state for less than 15 minutes, and in some cases, even less than 10 minutes. When an extruder is used to produce the molten feed, the times above refer to the mean time from when material is introduced to the extruder to when the molten mixture is congealed. Such mean times can be determined by procedures well known in the art. In one exemplary method, a small amount of dye or other similar compound is added to the feed while the extruder is operating under nominal conditions. Congealed multiparticulates are then collected over time and analyzed for the dye, from which the mean time is determined.
When the drug is a crystalline hydrate, it may be desirable to maintain a high water activity in the drug/glyceride/poloxamer admixture to reduce dehydration of the drug. This can be accomplished either by adding water to the powdered feed blend or by injecting water directly into the extruder by metering a controlled amount of water into a separate delivery port. In either case, sufficient water should be added to ensure the water activity is high enough to maintain the desired form of the crystalline
drug. Generally, it is desirable to keep the water activity of any material in contact with drug hydrate in the 30 percent to 100 percent RH range. This can be accomplished by ensuring that the concentration of water in the molten carrier is 30 percent to 100 percent of the solubility of water in the molten glyceride/poloxamer mixture at the maximum process temperature. In some cases, a small excess of water above the 100 percent water solubility limit may be added to the mixture.
Once the molten mixture has been formed, it is delivered to an atomizer that breaks the molten feed into small droplets. Virtually any method can be used to deliver the molten mixture to the atomizer, including the use of pumps and various types of pneumatic devices (e.g., pressurized vessels, piston pots). When an extruder is used to form the molten mixture, the extruder itself can be used to deliver the molten mixture to the atomizer. Typically, the molten mixture is maintained at an elevated temperature while delivering the mixture to the atomizer to prevent solidification of the mixture and to keep the molten mixture flowing.
Generally, atomization occurs in one of several ways, including (1 ) by
"pressure" or single-fluid nozzles; (2) by two-fluid nozzles; (3) by centrifugal or spinning-disk atomizers, (4) by ultrasonic nozzles; and (5) by mechanical vibrating nozzles. Detailed descriptions of atomization processes can be found in Lefebvre, Atomization and Sprays (1989) or in Perry's Chemical Engineers' Handbook (7th Ed. 1997). Preferably, a centrifugal or spinning-disk atomizer is used, such as the FX1 100-mm rotary atomizer manufactured by Niro A S (Soeborg, Denmark).
Once the molten mixture has been atomized, the droplets are congealed, typically by contact with a gas or liquid at a temperature below the solidification temperature of the droplets. Typically, it is desirable that the droplets are congealed in less than about 60 seconds, preferably in less than about 10 seconds, more preferably in less than about 1 second. Often, congealing at ambient temperature results in sufficiently rapid solidification of the droplets. However, the congealing step often occurs in an enclosed space to simplify collection of the multiparticulates. In such cases, the temperature of the congealing media (either gas or liquid) will increase over time as the droplets are introduced into the enclosed space, potentially effecting the formation of the multiparticulates or the chemical stability of the drug. Thus, a cooling gas or liquid is often circulated through the enclosed space to
maintain a constant congealing temperature. When it is desirable to minimize the time the drug is exposed to high temperatures, e.g., to prevent degradation, the cooling gas or liquid can be cooled to below ambient temperature to promote rapid congealing, thus minimizing formation of degradants.
Following formation of the multiparticulates, it may be desired to post-treat the multiparticulates to improve drug crystallinity and/or the stability of the multiparticulate.
The multiparticulates may also be mixed or blended with one or more pharmaceutically acceptable materials to form a suitable dosage form. Suitable dosage forms include tablets, capsules, sachets, oral powders for constitution, and the like.
Following formation of the melt spray congeal multiparticulates, the multiparticulates may optionally be coated with an additional exterior coating. The exterior coating may be any conventional coating, such as a protective film coating, a coating to provide delayed or sustained release of the drug, or to provide tastemasking.
In one embodiment, the coating is an enteric coating to provide delayed release of the drug. By "enteric coating" is meant an acid resistant coating that remains intact and does not dissolve at pH of less than about 4. The enteric coating surrounds the multiparticulate so that the solid amorphous dispersion layer does not dissolve or erode in the stomach. The enteric coating may include an enteric coating polymer. Enteric coating polymers are generally polyacids having a pKa of about 3 to 5. Examples of enteric coating polymers include: cellulose derivatives, such as cellulose acetate phthalate, cellulose acetate trimellitate, hydroxypropyl methyl cellulose acetate succinate, cellulose acetate succinate, carboxy methyl ethyl cellulose, methylcellulose phthalate, and ethylhydroxy cellulose phthalate; vinyl polymers, such as polyvinyl acetate phthalate, vinyl acetate-maleic anhydride copolymer; polyacrylates; and polymethacrylates such as methyl acrylate-methacrylic acid copolymer, methacrylate-methacrylic acid-octyl acrylate copolymer; and styrene- maleic mono-ester copolymer. These may be used either alone or in combination, or together with other polymers than those mentioned above.
One class of enteric coating materials are the pharmaceutically acceptable methacrylic acid copolymer which are copolymers, anionic in character, based on methacrylic acid and methyl methacrylate. Some of these polymers are known and sold as enteric polymers, for example having a solubility in aqueous media at pH 5.5 and above, such as the commercially available EUDRAGIT enteric polymers, such as Eudragit L 30, a polymer synthesized from dimethylaminoethyl methacrylate and Eudragit S and Eudragit FS.
The exterior coatings may include conventional plasticizers, including dibutyl phthalate; dibutyl sebacate; diethyl phthalate; dimethyl phthalate; triethyl citrate; benzyl benzoate; butyl and glycol esters of fatty acids; mineral oil; oleic acid; stearic acid; cetyl alcohol; stearyl alcohol; castor oil; corn oil; coconut oil; and camphor oil; and other excipients such as anti-tack agents, glidants, etc. For plasticizers, triethyl citrate, coconut oil and dibutyl sebacate are particularly preferred.
Exterior coatings can be formed using solvent-based and hot-melt coating processes. In solvent-based processes, the coating is made by first forming a solution or suspension comprising the solvent, the coating material and optional coating additives. The coating materials may be completely dissolved in the coating solvent, or only dispersed in the solvent as an emulsion or suspension or a combination of the two. Latex dispersions are an example of an emulsion or suspension that may be useful as in a solvent-based coating process. In one aspect, the solvent is a liquid at room temperature.
Coating may be conducted by conventional techniques, such as by pan coaters, rotary granulators and fluidized bed coaters such as top-spray, tangential- spray or bottom-spray (Wurster coating). A top-spray method can also be used to apply the coating. In this method, coating solution is sprayed down onto the fluidized cores. The solvent evaporates from the coated cores and the coated cores are re- fluidized in the apparatus. Coating continues until the desired coating thickness is achieved. Compositions and methods for making the multiparticulates of this embodiment are detailed in the following US Patent Applications, US 2005-0181062, US 2005-0181062, US 2008-0199527, US 2005-0186285A1 which are herein incorporated as reference in their entirety.
The multiparticulates of the invention generally are of a mean diameter from about 40 to about 3,000 micron, with a preferred range of 50 to 1 ,000 micron, and most preferably from about 100 to 300 micron. While the multiparticulates can have any shape and texture, it is preferred that they be spherical, with a smooth surface texture. These physical characteristics of the multiparticulates improve their flow properties, permit them to be uniformly coated (if desired). As used herein, the term "about" means +/- 10% of the value.
The multiparticulates of the present invention are particularly suitable for controlled release or delayed release or any combination of these two release profiles when introduced to a use environment. As used herein, a "use environment" can be either the in vivo environment of the gastrointestinal (Gl) tract or the in vitro dissolution tests described herein. Information about in vivo release rates can be determined from the pharmacokinetic profile using standard deconvolution or Wagner-Nelson treatment of the data which should be readily known to those skilled in the art.
Once the latrepirdine matrix multiparticulates are formed through methods described above, they may be blended with compressible excipients such as lactose, microcrystalline cellulose, dicalcium phosphate, and the like and the blend compressed to form a tablet. Disintegrants such as sodium starch glycolate or crosslinked polyvinyl pyrrolidone) are also usefully employed. Tablets prepared by this method disintegrate when placed in an aqueous medium (such as the Gl tract), thereby exposing the multiparticulate matrix which releases latrepirdine there from.
Other conventional formulation excipients may be employed in the controlled release portion of the invention, including those excipients well known in the art, e.g., as described in Remington: The Science and Practice of Pharmacy, 20th edition (2000). Generally, excipients such as surfactants, pH modifiers, fillers, matrix materials, complexing agents, solubilizers, pigments, lubricants, glidants, flavorants, and so forth may be used for customary purposes and in typical amounts without adversely affecting the properties of the compositions.
Example matrix materials, fillers, or diluents include lactose, mannitol, xylitol, dextrose, sucrose, sorbitol, compressible sugar, microcrystalline cellulose, powdered cellulose, starch, pregelatinized starch, dextrates, dextran, dextrin, dextrose,
maltodextrin, calcium carbonate, dibasic calcium phosphate, tribasic calcium phosphate, calcium sulfate, magnesium carbonate, magnesium oxide, poloxamers, polyethylene oxide, hydroxypropyl methyl cellulose and mixtures thereof.
In one embodiment, the controlled release portion comprises:
5 wt% to 60 wt% of latrepirdine
10 to 80 wt%, preferably 20 to 60 wt% matrix material;
2 to 45 wt%, preferably 15 to 35 wt% diluent; and
0.05 to 2 wt% lubricant.
The total amount of latrepirdine in the dosage form may range from 1 mg to 100 mg, preferably 15 mg to 85 mg.
Sustained Release - Osmotic Systems
In another embodiment, latrepirdine is incorporated into an osmotic delivery devices or "osmotic pumps" as they are known in the art. Osmotic pumps comprise a core containing an osmotically effective composition surrounded by a semipermeable membrane. The term "semipermeable" in this context means that water can readily diffuse through the membrane, but solutes dissolved in water typically cannot. In use, when placed in an aqueous environment, the device imbibes water due to the osmotic activity of the core composition. Owing to the semipermeable nature of the surrounding membrane, the contents of the device (including latrepirdine and any excipients) cannot pass through the non-porous regions of the membrane and are driven by osmotic pressure to leave the device through an opening or passageway pre-manufactured into the dosage form or, alternatively, formed in situ in the Gl tract as by the bursting of intentionally-incorporated weak points in the coating under the influence of osmotic pressure. The osmotically effective composition includes water- soluble species, which generate a colloidal osmotic pressure, and water-swellable polymers. Examples of such dosage forms are well known in the art. See, for example, Remington: The Science and Practice of Pharmacy, 21st Edition, 2006 Chapter 47; page 950-1 and herein incorporated as reference.
In one embodiment of the present invention, latrepirdine is incorporated into a bilayer osmotic delivery device such that the latrepirdine-containing composition must include an entraining agent in the form of a water-swellable polymer and a second push layer or water swelling layer which contains water-swellable polymers and/or
osmoticallly active agents, but does not contain any active agent. The bilayer tablet is surrounded by a semi-permeable membrane which contains one or more openings which are manufactured into the dosage form through such techniques as laser drilling. Such water-swellable polymers are often referred to in the pharmaceutical arts as an "osmopolymer" or a "hydrogel." The entraining agent suspends or entrains the drug so as to aid in the delivery of the drug through the delivery port(s). While not wishing to be bound by any particular theory, it is believed that upon the imbibition of water into the dosage form, the entraining agent has enough viscosity to allow it to suspend or entrain the drug, while at the same time remaining sufficiently fluid to allow the entraining agent to pass through the delivery port(s) along with the drug. The amount of the entraining agent present in the latrepirdine-containing composition may range from about 20 wt % to about 80 wt %. The entraining agent may be a single material or a mixture of materials. Non-crosslinked polyethylene oxide (PEO) may be used as the entraining agent. Other suitable entraining agents include hydroxypropyl cellulose (HPC), hydroxypropylmethyl cellulose (HPMC), methylcellulose (MC), hydroxyethyl cellulose (HEC) and polyvinyl pyrrolidone (PVP), as well as mixtures of these polymers with PEO.
The choice of the molecular weight for the PEO depends in part on whether the PEO makes up the bulk of the non-latrepirdine portion of the latrepirdine- containing composition, or whether significant amounts of other low-molecular weight water-soluble excipients are included; that is, the PEO molecular weight choice depends on the fraction of the latrepirdine-containing composition that is PEO. Should the latrepirdine-containing composition not become fluid rapidly, the dosage form can swell and rupture the coating that surrounds the core, potentially causing failure of the dosage form. Where the excipients of the latrepirdine-containing composition are primarily PEO (e.g., PEO makes up about 60 wt % or more of the non-latrepirdine components of the latrepirdine-containing composition), it is generally preferred that the PEO have an average molecular weight of from about 100,000 to 300,000 daltons. (As used herein, reference to molecular weights of polymers should be taken to mean average molecular weights.)
Alternatively, another embodiment of the present invention uses a higher molecular weight of PEO from about 500,000 to 800,000 daltons at a lower fraction of
the non-latrepirdine excipients, a portion of the PEO being replaced with a fluidizing agent. Ordinarily, when PEO makes up about 60 wt % or more of the non-latrepirdine components of the latrepirdine-containing composition, PEO having a molecular weight of 500,000 daltons or more makes the latrepirdine-containing composition too viscous, and can result in a rupture of the coating or at least in a delay of the release of latrepirdine. However, it has been found that such higher molecular weight PEO is preferred when the non-latrepirdine components of the latrepirdine-containing composition comprise less than about 60 wt % PEO and also contain a fluidizing agent. When using a higher molecular weight PEO, the amount of fluidizing agent present in the latrepirdine-containing composition may range from about 5 to about 50 wt %, preferably 10 to 30 wt % of the latrepirdine-containing composition. Preferred fluidizing agents are low molecular weight, water-soluble solutes such as non-reducing sugars and organic acids with aqueous solubilities of 30 img/mL or greater. Suitable sugars include xylitol, mannitol, sorbitol, and maltitol. Salts useful as a fluidizing agent include sodium chloride, sodium lactate and sodium acetate. Organic acids useful as a fluidizing agent include adipic acid, citric acid, malic acid, fumaric acid, succinic acid and tartaric acid.
The presence of the fluidizing agent, along with a relatively low level of higher molecular weight PEO (e.g., about 500,000 to about 800,000 daltons) allows the latrepirdine-containing composition to rapidly reach a low viscosity upon imbibition of water. In addition, it has been found that such an embodiment is capable of delivering relatively high amounts of latrepirdine.
The latrepirdine-containing composition may also contain other water- swellable polymers. For example, the latrepirdine-containing composition may contain relatively small amounts of water-swellable polymers that greatly expand in the presence of water. Such water-swellable polymers include sodium starch glycolate, sold under the trade name EXPLOTAB, and croscarmelose sodium, sold under the trade name AC-DI-SOL. Such polymers may be present in amounts ranging from 0 wt % to 10 wt % of the latrepirdine-containing composition.
The latrepirdine-containing composition may optionally include osmotically effective solutes, often referred to as "osmogens" or "osmagents." The amount of osmagent present in the latrepirdine-containing composition may range from about 0
wt % to about 50 wt %, preferably 10 wt % to 30 wt % of the latrepirdine-containing composition. Typical classes of suitable osmagents are water-soluble salts, sugars, organic acids, and other low-molecule-weight organic compounds that are capable of imbibing water to thereby establish an osmotic pressure gradient across the barrier of the surrounding coating. Typical useful salts include magnesium sulfate, magnesium chloride, calcium chloride, sodium chloride, lithium chloride, potassium sulfate, sodium carbonate, sodium sulfite, lithium sulfate, potassium chloride, and sodium sulfate. Conventionally, chloride salts such as sodium chloride are utilized as osmagents.
The latrepirdine-containing composition may further include solubility- enhancing agents or solubilizers that promote the aqueous solubility of the drug, present in an amount ranging from about 0 to about 30 wt % of the latrepirdine- containing composition. Solubilizers useful with latrepirdine include organic acids and organic acid salts, partial glycerides, e.g., less than fully esterified derivatives of glycerin, including glycerides, monoglycerides, diglycerides, glyceride derivatives, polyethylene glycol esters, polypropylene glycol esters, polyhydric alcohol esters, polyoxyethylene ethers, sorbitan esters, polyoxyethylene sorbitan esters, and carbonate salts.
A preferred class of solubilizers is organic acids. Since latrepirdine is a base which is solubilized by protonation, and since its solubility in an aqueous environment of pH 5 or higher is reduced, it is believed that addition of an organic acid to the Latrepirdine-containing composition assists in solubilization and hence absorption of latrepirdine. Even a slight decrease in the pH of the aqueous solution at high pH results in dramatic increases in the solubility of latrepirdine. Organic acids can also promote stability during storage prior to introduction to a use environment due to their tendency to maintain latrepirdine in a protonated state.
There are a variety of factors to consider when choosing an appropriate organic acid for use as a solubilizer with latrepirdine in an osmotic dosage form. The acid should not interact adversely with latrepirdine, should have appropriate water solubility, and should provide good manufacturing properties.
Accordingly, it has been found that a preferred subset of organic acids meeting such criteria consists of citric, succinic, fumaric, adipic, malic and tartaric
acids. Citric, malic, and tartaric acid have the advantage of high water solubility and high osmotic pressure. Succinic and fumaric acid offer a combination of both moderate solubility and moderate osmotic pressure.
The water-swellable composition may also optionally contain a colorant. The purpose of the colorant is to allow identification of the drug-containing side of the tablet face for purposes of providing the delivery port, such as by laser drilling through the coating. Acceptable colorants include, but are not limited to, Red Lake No. 40, FD C Blue 2 and FD C Yellow 6.
Both the Latrepirdine-containing composition and/or the water-swellable composition may optionally contain an antioxidant, such as but not limited to BHT, vitamin E, BHA, or ascorbyl palmitate. The antioxidant may be present in an amount ranging from 0 to 1 wt % of the Latrepirdine-containing and/or the water-swellable composition.
Water-swellable composition may also include other conventional pharmaceutically useful excipients such as a binder, including HPC, HPMC, HEC, MC, and PVP, a tableting aid, such as microcrystalline cellulose, and a lubricant such as magnesium stearate.
The water-swellable composition is prepared by mixing the water-swellable polymer and the other excipients to form a uniform blend. To obtain a uniform blend, it is desirable to either wet or dry granulate or dry blend ingredients that have similar particle sizes using the types of processes known to those skilled in the art.
TABLETING
The core is prepared by first placing a mixture of the latrepirdine-containing composition into a tablet press and then leveling the mixture by gentle compression. The water-swellable composition is then placed on top of the latrepirdine-containing composition and compressed in order to complete formation of the core. Alternatively, the water-swellable composition can be placed into the tablet press first, followed by the latrepirdine-containing composition.
The respective amounts of latrepirdine-containing composition and water- swellable composition are chosen to provide satisfactory latrepirdine release. When it is desired to provide a large latrepirdine dose in a relatively small dosage size, it is desired to maximize the amount of latrepirdine-containing composition and minimize
the amount of water-swellable composition, while still obtaining good release performance. In the dosage forms of the present invention, when the water-swellable polymer in the water-swellable composition is only PEO, the latrepirdine-containing composition may comprise from about 50 to about 85 wt % of the core, and preferably from about 60 to about 70 wt %. These values correspond to a weight ratio of the latrepirdine-containing composition to water-swellable composition of from 1 to about 5.7. When all or part of the water-swellable polymer in the water-swellable composition comprises sodium starch glycolate or croscarmellose sodium, the latrepirdine-containing composition may comprise from 50 to 90 wt % of the core, and preferably from about 75 to about 85 wt %. Those values correspond to the weight ratio of the latrepirdine-containing composition to water-swellable composition of from 1 to 9. The absolute value of the diameter and height of the tablets of the present invention can vary over a wide range.
THE COATING
Following formation of the core, the semi-permeable coating is applied. The coating should have high water permeability and a high strength, while at the same time be easily fabricated and applied. High water permeability is required to permit water to enter the core in sufficient volume. High strength is required to ensure the coating does not burst when the core swells as it imbibes water, leading to an uncontrolled delivery of the core contents. Finally, the coating must have high reproducibility and yield.
It is essential that the coating have at least one delivery port in communication with the interior and exterior of the coating for delivery of the latrepirdine-containing composition. Furthermore, the coating must be non-dissolving and non-eroding during release of the latrepirdine-containing composition, generally meaning that it be water-insoluble, such that latrepirdine is substantially entirely delivered through the delivery port(s), in contrast to delivery via permeation through the coating.
Coatings with these characteristics can be obtained using hydrophilic polymers such as plasticized and unplasticized cellulose esters, ethers, and ester- ethers. Particularly suitable polymers include cellulose acetate (CA), cellulose acetate butyrate (CAB), and ethyl cellulose (EC). One set of polymers are cellulose
acetates having acetyl contents of 25 to 42%. One typical polymer is CA having an acetyl content of 39.8%, specifically, CA 398-10 (Eastman Fine Chemicals, Kingsport, Tenn.). CA 398-10 is reported to have an average molecular weight of about 40,000 daltons. Another typical CA having an acetyl content of 39.8% is high molecular weight CA having an average molecular weight greater than about 45,000, and specifically, CA 398-30 (Eastman Fine Chemical) which is reported to have an average molecular weight of 50,000 daltons.
Coating is conducted in conventional fashion by first forming a coating solution and then coating by dipping, fluidized bed coating, or by pan coating. To accomplish this, a coating solution is formed comprising the polymer and a solvent. Typical solvents useful with the cellulosic polymers above include acetone, methyl acetate, ethyl acetate, isopropyl acetate, n-butyl acetate, methyl isobutyl ketone, methyl propyl ketone, ethylene glycol monoethyl ether, ethylene glycol monoethyl acetate, methylene dichloride, ethylene dichloride, propylene dichloride, nitroethane, nitropropane, tetrachloroethane, 1 ,4-dioxane, tetrahydrofuran, diglyme, and mixtures thereof. The coating solution typically contains 3 to 15 wt % of the polymer.
The coating solution may also include pore-formers or non-solvents in any amount as long as the polymer remains soluble at the conditions used to form the coating and as long as the coating remains water permeable and has sufficient strength. Pore-formers and their use in fabricating coatings are described in U.S. Pat. Nos. 5,698,220 and 5,612,059, the pertinent disclosures of which are incorporated herein by reference. The term "pore former," as used herein, refers to a material added to the coating solution that has low or no volatility relative to the solvent such that it remains as part of the coating following the coating process but that is sufficiently water swellable or water soluble such that, in the aqueous use environment it provides a water-filled or water-swollen channel or "pore" to allow the passage of water, thereby enhancing the water permeability of the coating. Suitable pore formers include but are not limited to hydroxypropylcellulose (HPC), polyethylene glycol ("PEG"), PVP, and PEO. To obtain a combination of high water permeability and high strength when PEG or HPC are used as a pore former, the weight ratio of CA:PEG or CA:HPC should range from about 6:4 to about 9:1 .
The addition of a non-solvent such as water to the coating solution results in exceptional performance. By "non-solvent" is meant any material added to the coating solution that substantially dissolves in the coating solution and reduces the solubility of the coating polymer or polymers in the solvent. In general, the function of the non-solvent is to impart porosity to the resulting coating. As described below, porous coatings have higher water permeability than an equivalent weight of a coating of the same composition that is not porous and this porosity is indicated by a reduction in the density of the coating (mass/volume). Although not wishing to be bound by any particular mechanism of pore formation, it is generally believed that addition of a non-solvent imparts porosity to the coating during evaporation of solvent by causing the coating solution to undergo liquid and liquid phase separation prior to solidification. The suitability and amount of a particular candidate material can be evaluated for use as a non-solvent by progressively adding the candidate non- solvent to the coating solution until it becomes cloudy. If this does not occur at any addition level up to about 50 wt % of the coating solution, it generally is not appropriate for use as a non-solvent. When clouding is observed, termed the "cloud point," an appropriate level of non-solvent for maximum porosity is the amount just below the cloud point. For acetone solutions comprising 7 wt % CA and 3 wt % PEG, the cloud point is at about 23 wt % water. When lower porosities are desired, the amount of non-solvent can be reduced as low as desired.
Suitable non-solvents are any materials that have appreciable solubility in the solvent and that lower the coating polymer solubility in the solvent. The preferred non-solvent depends on the solvent and the coating polymer chosen. In the case of using a volatile polar coating solvent such as acetone, suitable non-solvents include water, glycerol, alcohols such as methanol or ethanol.
When using CA 398-10, coating solution weight ratios of CA:PEG 3350: water are 7:3:5, 8:2:5, and 9:1 :5, with the remainder of the solution comprising a solvent such as acetone. Thus, for example, in a solution having a weight ratio of CA:PEG 3350: water of 7:3:5, CA comprises 7 wt % of the solution, PEG 3350 comprises 3 wt % of the solution, water comprises 5 wt % of the solution, and acetone comprises the remaining 85 wt %.
Coatings formed from these coating solutions are generally porous. By "porous" is meant that the coating in the dry state has a density less than the density of the same material in a nonporous form. By "nonporous form" is meant a coating material formed by using a coating solution containing no non-solvent, or the minimal amount of non-solvent required to produce a homogeneous coating solution. The dry-state density of the coating can be calculated by dividing the coating weight (determined from the weight gain of the tablets before and after coating) by the coating volume (calculated by multiplying the coating thickness, as determined by optical or scanning electron microscopy, by the tablet surface area). The porosity of the coating is one of the factors that leads to the combination of high water permeability and high strength of the coating.
The weight of the coating around the core depends on the composition and porosity of the coating, but generally should be present in an amount ranging from 3 to 30 wt %, based on the weight of the uncoated core. A coating weight of at least about 8 wt %, is typically preferred for sufficient strength for reliable performance.
While porous coatings based on CA, PEG, and water described above translate to excellent results, other pharmaceutically acceptable materials could be used in the coating so long as the coating has the requisite combination of high water permeability, high strength, and ease of fabrication and application. Further, such coatings may be dense, porous, or "asymmetric," having one or more dense layers and one or more porous layers such as those disclosed in U.S. Pat. Nos. 5,612,059 and 5,698,220, the pertinent disclosures of which are incorporated herein by reference.
The coating must also contain at least one delivery port in communication with the interior and exterior of the coating to allow for release of the drug-containing composition to the exterior of the dosage form. The delivery port can range in size from about the size of the drug particles, and thus could be as small as 1 to 100 microns in diameter and may be termed pores, up to about 5000 microns in diameter. The shape of the port may be substantially circular, in the form of a slit, or other convenient shape to ease manufacturing and processing. The port(s) may be formed by post-coating mechanical or thermal means or with a beam of light (e.g., a laser), a beam of particles, or other high-energy source, or may be formed in situ by rupture of
a small portion of the coating. Such rupture may be controlled by intentionally incorporating a relatively small weak portion into the coating. Delivery ports may also be formed in situ by erosion of a plug of water-soluble material or by rupture of a thinner portion of the coating over an indentation in the core. Delivery ports may be formed by coating the core such that one or more small regions remains uncoated. In addition, the delivery port can be a large number of holes or pores that may be formed during coating, as in the case of asymmetric membrane coatings, described in more detail herein, and of the type disclosed in U.S. Pat. Nos. 5,612,059 and 5,698,220, the disclosures of which are incorporated by reference. When the delivery pathways are pores there can be a multitude of such pores that range in size from 1 micron to greater than 100 microns. During operation, one or more of such pores may enlarge under the influence of the hydrostatic pressure generated during operation. At least one delivery port should be formed on the side of coating that is adjacent to the latrepirdine-containing composition, so that the latrepirdine-containing composition will be extruded out of the delivery port by the swelling action of the water-swellable composition. It is recognized that some processes for forming delivery ports may also form holes or pores in the coating adjacent to the water- swellable composition.
The coating may optionally include a port in communication with the water- swellable composition. Such a delivery port does not typically alter the latrepirdine release characteristics of the dosage form, but may provide manufacturing advantages. It is believed that the water-swellable compositions, such as those containing PEO with a molecular weight between 3,000,000 and 8,000,000 daltons, are too viscous to appreciably exit the port. In dosage forms wherein the delivery ports are drilled either mechanically or by laser, the tablet must be oriented so that at least one delivery port is formed in the coating adjacent to the latrepirdine-containing composition. A colorant within the water-swellable composition is used to orient the core dosage form during the drilling step in manufacture. By providing a delivery port on both faces of the dosage form, the need to orient the dosage form may be eliminated and the colorant may be removed from the water-swellable composition.
In yet another embodiment, latrepirdine is incorporated into a variation of the above disclosed osmotic delivery device, an asymmetric membrane technology
(AMT). These devices have been disclosed in Herbig, et al., J. Controlled Release, 35, 1995, 127-136, and U.S. Pat. Nos. 5,612,059 and 5,698,220 as coatings in osmotic drug delivery systems. These AMT systems provide the general advantages of osmotic controlled release devices (reliable drug delivery independent of position in gastrointestinal tract), yet do not require the added manufacturing step of drilling a hole in the coating, as seen with a number of other osmotic systems. In the formation of these porous coatings, a water-insoluble polymer is combined with a water- soluble, pore-forming material. The mixture is coated onto an osmotic tablet core from a combination of water and solvent. As the coating dries, a phase inversion process occurs whereby a porous, asymmetric membrane is produced. The use of an AMT system for controlled release of a drug with similar physiochemical properties is described in US Patent Application Publication US2007/0248671 and herein incorporated as reference.
While a number of materials have been disclosed for use as pore-formers in the production of asymmetric membranes, the previously disclosed materials all bring chemical or physical stability issues into the system. In particular, many of the prior art materials are liquids, which can potentially migrate out of the coating during storage. Of the ones that are solid, both polymeric materials and inorganic materials have been taught. Inorganic materials can be difficult to use for a number of reasons. In particular, they often have a tendency to crystallize and/or adsorb moisture on storage. The particular polymeric materials that have been taught include polyvinylpyrrolidone (PVP) and polyethylene glycol (PEG) derivatives. Both of these materials have a strong tendency to form peroxides and/or formaldehyde upon storage (see for example Waterman, et al., "Impurities in Drug Products" in Handbook of Isolation and Characterization of Impurities in Pharmaceuticals, S. Ajira and K. M. Alsante, Eds. 2003, pp. 75-85). Many drug substances are reactive with such polymer degradation products, both because of their intrinsic reactivity and their tendency to migrate upon storage. However, this formulation space is relatively narrow. U.S. Pat. No. 4,519,801 discloses a wide list of water-soluble polymeric components useful for coatings in osmotic systems, but fails to teach appropriate selections of water-soluble components for AMT systems. There remains, therefore, a need for new pore-forming materials for AMT systems wherein the pore-forming
materials do not generate reactive byproducts, crystallize or migrate from the coating upon storage.
One aspect of the present invention provides a dosage form which comprises (a) a core containing at least one pharmaceutically active ingredient and (b) at least one asymmetric membrane technology coating wherein said coating comprises: a. one or more substantially water-insoluble polymers, and
b. one or more solid, water-soluble polymeric materials that do not contain amounts of hydrogen peroxide or formaldehyde greater than about 0.01 percent w:w after storage at 40 degrees C./75 percent RH for 12 weeks.
One aspect of the present invention also provides a dosage form wherein the dosage form delivers drug primarily by osmotic pressure. In particular embodiments, the present invention provides a dosage form wherein the pharmaceutically active ingredient is latrepirdine or a pharmaceutically acceptable salt thereof. The water- insoluble polymer as used in the present invention preferably comprises a cellulose derivative, more preferably, cellulose acetate. The solid, water-soluble polymeric material as used in the present invention comprises a polymer having a weight average molecular weight between 2000 and 50,000 daltons. In preferable embodiments, the solid, water-soluble polymeric material is selected from the group consisting of water-soluble cellulose derivatives, acacia, dextrin, guar gum, maltodextrin, sodium alginate, starch, polyacrylates, polyvinyl alcohols and zein. In particular embodiments, the water-soluble cellulose derivatives comprise hydroxypropylcellulose, hydroxypropylmethylcellulose and hydroxyethylcellulose. In certain embodiments, the solid, water-soluble, polymeric material has a viscosity for a 5 percent w:w aqueous solution of less than 400 mPa s. In certain other embodiments, the solid, water-soluble, polymeric material has a viscosity for a 5 percent w:w aqueous solution of less than 300 mPa s. In other embodiments, the solid, water-soluble, polymeric material has a softening temperature greater than 55 degrees C.
The dosage form of the present invention may be a tablet or a multiparticulate. In certain embodiments, the core of the present invention contains a sugar. More preferably, the sugar is mannitol. In certain embodiments, the water- insoluble polymer is cellulose acetate and said solid, water-soluble polymeric
material is hydroxypropylcellulose. In certain preferred embodiments, the dosage form of the invention contains latrepirdine, or a pharmaceutically acceptable salt thereof, as the pharmaceutically active ingredient, while the water-insoluble polymer is cellulose acetate and the solid, water-soluble polymeric material is hydroxypropylcellulose.
A process of the present invention encompasses the process wherein the coating is applied from a mixture of acetone and water using a pan coating. The process of the present invention also encompasses the process wherein the asymmetric membrane comprises cellulose acetate and hydroxypropylcellulose which is coated from a mixture of acetone to water between about 9:1 and 6:4, w:w, and more preferably between about 3: 1 and about 4.5: 1 , w:w, using a pan coater. In particular, the process of the present invention encompasses the process wherein the core comprises latrepirdine, or a pharmaceutically acceptable salt thereof.
In the preparation of the asymmetric membrane coatings of the present invention, the water-insoluble component of the asymmetric membrane coating preferentially is formed from cellulose derivatives. In particular, these derivatives include cellulose esters and ethers, namely the mono-, di- and triacyl esters wherein the acyl group consists of two to four carbon atoms and lower alkyl ethers of cellulose wherein the alkyl group has one to four carbon atoms. The cellulose esters can also be mixed esters, such as cellulose acetate butyrate, or a blend of cellulose esters. The same variations can be found in ethers of cellulose and include blends of cellulose esters and cellulose ethers. Other cellulose derivatives which can be used in making asymmetric membranes of the present invention include cellulose nitrate, acetaldehyde dimethyl cellulose, cellulose acetate ethyl carbamate, cellulose acetate phthalate, cellulose acetate methyl carbamate, cellulose acetate succinate, cellulose acetate dimethaminoacetate, cellulose acetate ethyl carbonate, cellulose acetate dimethaminoacetate, cellulose acetate ethyl carbonate, cellulose acetate chloroacetate, cellulose acetate ethyl oxalate, cellulose acetate methyl sulfonate, cellulose acetate butyl sulfonate, cellulose acetate p-toluene sulfonate, cellulose cyanoacetates, cellulose acetate trimellitate, cellulose methacrylates and hydroxypropylmethylcellulose acetate succinate. A particularly preferred water- insoluble component is cellulose acetate. Particularly preferred cellulose acetates
include those having an acetyl content of about 40 percent and a hydroxyl content of about 3.5 percent. Other materials also can be used in the fabrication of asymmetric membrane technology coatings, provided such materials are substantially water- insoluble, film-forming and safe to use in pharmaceutical applications.
In the preparation of the asymmetric membrane coatings of the present invention, yhe water-soluble polymeric component of the present invention comprises solid, polymeric materials that do not form hydrogen peroxide or formaldehyde upon storage for 12 weeks at 40 degrees C./75 percent relative humidity, in an amount greater than about 0.01 percent w/w (100 parts per million, ppm). In terms of water solubility, the solid polymeric water-soluble material preferentially has a water- solubility of greater than 0.5 img/mL; more preferably, greater than 2 img/mL; and still more preferably, greater than 5 img/mL
The solid polymeric water-soluble material has a melting or softening temperature above room temperature. Preferentially, the solid material has a melting or softening temperature above 30 degrees C; more preferentially, above 40 degrees C; and most preferentially, above 50 degrees C. Melting and softening points can be determined visually using a melting point apparatus, or alternatively, can be measured using differential scanning calorimetry (DSC), as is known in the art. The polymer can be either a homopolymer or a copolymer. Such polymers can be natural polymers, or be derivatives of natural products, or be entirely synthetic. The molecular weight of such materials is preferentially high enough to prevent migration and aid in film-forming, yet low enough to allow coating (as discussed below). The preferred molecular weight range for the present invention is therefore between 2000 and 50,000 daltons (weight average). Preferred polymers suitable as water-soluble components of an asymmetric membrane technology coating for the present invention include substituted, water-soluble cellulose derivatives, acacia, dextrin, guar gum, maltodextrin, sodium alginate, starch, polyacrylates, polyvinyl alcohols and zein. Particularly preferred water-soluble polymers include hydroxyethylcellulose, hydroxypropylcellulose and polyvinylalcohol.
It is difficult to obtain asymmetric membrane coatings if the viscosity of the coating solution is too high, and that one approach to solving this issue is to use more dilute solutions of the polymer. Due to the phase behavior of the coating
solution, having both water-soluble and organic-soluble components, there is a limit to how low the concentration of the water-soluble polymer can be and still provide a commercializable process. For this reason, it is preferred that the water-soluble polymers not have too high a viscosity. Viscosities can be determined at 25 degrees C. using a Brookfield LVF viscometer (available from Brookfield Engineering Corp., Middleboro, Mass.) with spindle and speed combinations depending on viscosity levels for 5 percent (w:w) aqueous solutions. Preferred water-soluble polymers have viscosities for 5 percent (w:w) solutions of less than 400 mPa s; more preferably, less than 300 mPa s.
Using the above criteria, especially preferred water-soluble polymers include hydroxypropylcellulose and hydroxyethylcellulose having a viscosity for a 5 percent (w:w) of less than 300 mPa s. Commercially available examples of such polymers include Klucel EF.TM. and Natrasol LR.TM., both made by the Aqualon Division of Hercules Corp., Hopewell, Va.
The water-soluble, solid polymeric material's stability to formation of hydrogen peroxide can be measured by storing the polymer in an oven having a temperature and relative humidity (RH) of 40 degrees C. and 75 percent RH, respectively. The polymer should be stored exposed to the oven environment under "open" conditions. The polymer should be stored for at least 12 weeks. Levels of hydrogen peroxide can be administered as described in G. M. Eisenberg, "Colorimetric determination of hydrogen peroxide" in Ind. Eng. Chem. (Anal. Ed.), 1943, 15, 327-328. Under these storage conditions, acceptable polymeric materials for the present invention have hydrogen peroxide levels below 100 parts per million (ppm); more preferably, below 50 ppm; and most preferably, below 10 ppm.
Similarly, the water-soluble polymer's stability to formation of formaldehyde can be measured by storing the polymer in an oven at 40 degrees C. and 75 percent RH. Polymer should be stored in a sealed container to avoid loss of volatile formaldehyde. The polymer should be stored for at least 12 weeks. Levels of formaldehyde can be determined as described in M. Ashraf-Khorassani, et al., "Purification of pharmaceutical excipients with supercritical fluid extraction" in Pharm. Dev. Tech. 2005, 10, 1-10. Under these storage conditions, acceptable water-soluble
polymeric materials for the present invention have formaldehyde levels below 100 ppm, more preferably, below 50 ppm, and most preferably, below 10 ppm.
It will be appreciated by those skilled in the art that the asymmetric membrane technology coating formulation can contain small amounts of other materials without significantly changing its function or altering the nature of the present invention. Such additives include glidants (e.g., talc and silica) and plasticizers (e.g., triethylcitrate and triacetin), which are typically added, when needed, at levels of less than about 5 percent (w:w) of the coating.
It will be appreciated by those skilled in the art that active pharmaceutical ingredients can also be in the form of pharmaceutically acceptable salts. The cores for the present invention can also employ solubilizing additives. Such additives include pH-buffering additives to maintain the core at a pH wherein the active pharmaceutical ingredient has a sufficiently high solubility to be pumped out of the dosage form in solution. The active pharmaceutical ingredient can be present in the core at levels ranging from about 0.1 percent (w:w) to about 75 percent (w:w).
The core can contain osmotic agents which help to provide the driving force for drug delivery. Such osmotic agents include water-soluble sugars and salts. A particularly preferred osmotic agent is mannitol or sodium chloride.
The core of the AMT system can contain other additives to provide for such benefits as stability, manufacturability and system performance. Stabilizing excipients include pH-modifying ingredients, antioxidants, chelating agents, and other such additives as is known in the art. Excipients that improve manufacturability include agents to help in flow, compression or extrusion. Flow can be helped by such additives as talc, stearates and silica. Flow is also improved by granulation of the drug and excipients, as is known in the art. Such granulations often benefit from the addition of binders such as hydroxypropylcellulose, starch and polyvinylpyrollidone (povidone). Compression can be improved by the addition of diluents to the formulation. Examples of diluents include lactose, mannitol, microcrystalline cellulose and the like, as is known in the art. For cores produced by extrusion, the melt properties of the excipients can be important. Generally, it is preferable that such excipients have melting temperatures below about 100 degrees C. Examples of appropriate excipients for melt processes include esterified glycerines and stearyl
alcohol. For compressed dosage forms, manufacturability can be improved by addition of lubricants. A particularly preferred lubricant is magnesium stearate.
Cores can be produced using standard tablet compression processes, as is known in the art. Such processes involve powders filling dies followed by compression using appropriate punches. Cores can also be produced by an extrusion process. Extrusion processes are especially well-suited to making small cores (multiparticulates). A preferred extrusion process is a melt-spray-congeal process as described in WO2005/053653A1 , incorporated by reference. Cores can also be prepared by layering drug onto seed cores. Such seed cores are preferentially made of sugar or microcrystalline cellulose. Drug can be applied onto the cores by spraying, preferentially in a fluid-bed operation, as is known in the art.
In the practice of the subject invention, the cores are coated with the asymmetric membrane by any technique that can provide the asymmetric membrane as a coating over the entire cores. Preferred coating methods include pan coating and fluid-bed coating. In both coating processes, the water-insoluble polymer and water-soluble polymer as well as any other additives are first dissolved or dispersed in an appropriate solvent or solvent combination. In order to achieve a suitably porous membrane, the coating solvent needs to be optimized for performance. Generally, the solvents are chosen such that the more volatile solvent is the better solvent for the water-insoluble polymeric component. The result is that during coating, the water-insoluble polymeric component precipitates from solution. Preferred solvents and solvent ratios can be determined by examining the multi- component solubility behavior of the system. A preferred solvent mixture is acetone and water, with a ratio of between about 9: 1 and about 6:4, w:w.
Sustained Release - Reservoir Systems
Another class of latrepirdine sustained-release dosage forms of this invention includes membrane-moderated or reservoir systems. In this class, a reservoir of latrepirdine is surrounded by a rate-limiting membrane. The latrepirdine traverses the membrane by mass transport mechanisms well known in the art, including but not limited to dissolution in the membrane followed by diffusion across the membrane or diffusion through liquid-filled pores within the membrane. These individual reservoir system dosage forms may be large, as in the case of a tablet containing a single
large reservoir, or multiparticulate, as in the case of a capsule containing a plurality of reservoir particles, each individually coated with a membrane. The coating can be non-porous, yet permeable to latrepirdine (for example latrepirdine may diffuse directly through the membrane), or it may be porous. As with other embodiments of this invention, the particular mechanism of transport is not believed to be critical.
Sustained release coatings as known in the art may be employed to fabricate the membrane, especially polymer coatings, such as a cellulose ester or ether, an acrylic polymer, or a mixture of polymers. Preferred materials include ethyl cellulose, cellulose acetate and cellulose acetate butyrate. The polymer may be applied as a solution in an organic solvent or as an aqueous dispersion or latex. The coating operation may be conducted in standard equipment such as a fluid bed coater, a Wurster coater, or a rotary bed coater.
If desired, the permeability of the coating may be adjusted by blending of two or more materials. A useful process for tailoring the porosity of the coating comprises adding a pre-determined amount of a finely-divided water-soluble material, such as sugars or salts or water-soluble polymers to a solution or dispersion (e.g., an aqueous latex) of the membrane-forming polymer to be used. When the dosage form is ingested into the aqueous medium of the Gl tract, these water soluble membrane additives are leached out of the membrane, leaving pores which facilitate release of the drug. The membrane coating can also be modified by the addition of plasticizers, as known in the art.
A useful variation of the process for applying a membrane coating comprises dissolving the coating polymer in a mixture of solvents chosen such that as the coating dries, a phase inversion takes place in the applied coating solution, resulting in a membrane with a porous structure. Numerous examples of this type of coating system are given in European Patent Specification 0 357 369 B1 , published Mar. 7, 1990, herein incorporated by reference.
The morphology of the membrane is not of critical importance so long as the permeability characteristics enumerated herein are met. The membrane can be amorphous or crystalline. It can have any category of morphology produced by any particular process and can be, for example, an interfacially-polymerized membrane (which comprises a thin rate-limiting skin on a porous support), a porous hydrophilic
membrane, a porous hydrophobic membrane, a hydrogel membrane, an ionic membrane, and other such materials which are characterized by controlled permeability to latrepirdine.
A useful reservoir system embodiment is a capsule having a shell comprising the material of the rate-limiting membrane, including any of the membrane materials previously discussed, and filled with a latrepirdine drug composition. A particular advantage of this configuration is that the capsule may be prepared independently of the drug composition, thus process conditions that would adversely affect the drug can be used to prepare the capsule. One embodiment is a capsule having a shell made of a porous or a permeable polymer made by a thermal forming process. Another embodiment is a capsule shell in the form of an asymmetric membrane; e.g., a membrane that has a thin skin on one surface and most of whose thickness is constituted of a highly permeable porous material. A process for preparation of asymmetric membrane capsules comprises a solvent exchange phase inversion, wherein a solution of polymer, coated on a capsule-shaped mold, is induced to phase-separate by exchanging the solvent with a-miscible non-solvent. Examples of asymmetric membranes useful in this invention are disclosed in the aforementioned European Patent Specification 0 357 369 B1.
Another embodiment of the class of reservoir systems comprises a multiparticulate wherein each particle is coated with a polymer designed to yield sustained release of latrepirdine. The multiparticulate particles each comprise latrepirdine and one or more excipients as needed for fabrication and performance. The size of individual particles, as previously mentioned, is generally between about 50 micron and about 3 mm, although beads of a size outside this range may also be useful. In general, the beads comprise latrepirdine and one or more binders. As it is generally desirable to produce dosage forms which are small and easy to swallow, beads which contain a high fraction of latrepirdine relative to excipients are preferred. Binders useful in fabrication of these beads include microcrystalline cellulose (e.g., Avicel.RTM., FMC Corp.), hydroxypropyl cellulose (HPC), hydroxypropyl methyl cellulose (HPMC), and related materials or combinations thereof. In general, binders which are useful in granulation and tabletting, such as starch, pregelatinized starch, and poly (N-vinyl-2-pyrrolidinone) (PVP) may also be used to form multiparticulates.
Reservoir system latrepirdine multiparticulates may be prepared using techniques known to those skilled in the art, including, but not limited to, the techniques of extrusion and spheronization, wet granulation, fluid bed granulation, and rotary bed granulation. In addition, the beads may also be prepared by building the latrepirdine composition (drug plus excipients) up on a seed core (such as a nonpareil seed) by a drug-layering technique such as powder coating or by applying the latrepirdine composition by spraying a solution or dispersion of latrepirdine in an appropriate binder solution onto seed cores in a fluidized bed such as a Wurster coater or a rotary processor. An example of a suitable composition and method is to spray a dispersion of a latrepirdine/hydroxypropylcellulose composition in water. Advantageously, latrepirdine can be loaded in the aqueous composition beyond its solubility limit in water.
A method for manufacturing the multiparticulate cores of this embodiment is the extrusion/spheronization process, as previously discussed for matrix multiparticulates. Another process and composition for this method involves using water to wet-mass blend of about 5 to 75% of micro-crystalline cellulose with correspondingly about 95 to 25% latrepirdine. In another embodiment, the process involves the use of water to wet-mass blend of about 5-30% microcrystalline cellulose with correspondingly about 5-70% latrepirdine.
A sustained release coating as known in the art, especially polymer coatings, may be employed to fabricate the membrane, as previously discussed for reservoir systems. Suitable and preferred polymer coating materials, equipment, and coating methods also include those previously discussed.
The rate of latrepirdine release from the coated multiparticulates can also be controlled by factors such as the composition and binder content of the drug- containing core, the thickness and permeability of the coating, and the surface-to- volume ratio of the multiparticulates. It will be appreciated by those skilled in the art that increasing the thickness of the coating will decrease the release rate, whereas increasing the permeability of the coating or the surface-to-volume ratio of the multiparticulates will increase the release rate. If desired, the permeability of the coating may be adjusted by blending of two or more materials. A useful series of coatings comprises mixtures of water-insoluble and water-soluble polymers, for
example, ethylcellulose and hydroxypropyl methylcellulose, respectively. A useful modification to the coating is the addition of finely-divided water-soluble material, such as sugars or salts. When placed in an aqueous medium, these water soluble membrane additives are leached out of the membrane, leaving pores which facilitate delivery of the drug. The membrane coating may also be modified by the addition of plasticizers, as is known to those skilled in the art. Another useful variation of the membrane coating utilizes a mixture of solvents chosen such that as the coating dries, a phase inversion takes place in the applied coating solution, resulting in a membrane with a porous structure.
Another embodiment is a multiparticulate comprising about 5-50 % latrepirdine, the individual particles being coated with an aqueous dispersion of ethyl cellulose, which dries to form a continuous film.
Another embodiment is obtained when the latrepirdine beads are less than about 400 micron in size and are coated with a phase inversion membrane of ethyl cellulose or cellulose acetate.
Another embodiment is obtained when the latrepirdine beads are less than about 400 micron in size and are coated with an aqueous dispersion of ethyl cellulose, which dries to form a continuous film.
Another embodiment is obtained when the latrepirdine beads are less than about 300 micron in size and are coated with film an aqueous dispersion of ethyl cellulose, which dries to form a continuous film.
Delayed Release and Controlled Release Components
As it is an object of this invention to reduce the exposure of the upper Gl tract to high concentrations of latrepirdine, another class of dosage forms includes those forms which incorporate a delay before the onset of controlled release of latrepirdine. One embodiment can be illustrated by a tablet comprising a core containing latrepirdine coated with a first coating of a polymeric material of the type useful for controlled release of latrepirdine and a second coating of the type useful for delaying release of drugs when the dosage form is ingested. The first coating is applied over and surrounds the tablet. The second coating is applied over and surrounds the first coating.
The tablet can be prepared by techniques well known in the art and contains a therapeutically useful amount of latrepirdine plus such excipients as are necessary to form the tablet by such techniques.
The first coating may be a controlled release coating as known in the art, especially polymer coatings, to fabricate the membrane, as previously discussed for reservoir systems. Suitable polymer coating materials, equipment, and coating methods also include those previously discussed.
Materials useful for preparing the second coating on the tablet include polymers known in the art as enteric coatings for delayed-release of pharmaceuticals. These most commonly are pH-sensitive materials such as cellulose acetate phthalate, cellulose acetate trimellitate, hydroxypropyl methyl cellulose phthalate, polyvinyl acetate phthalate), and acrylic copolymers such as Eudragit L- 100 (RohmPharma), Eudragit L 30 D-55, Eudragit S 100, Eudragit FS 30D, and related materials, as more fully detailed below under "Delayed Release". The thickness and type of the delayed-release coating is adjusted to give the desired delay property. In general, thicker coatings are more resistant to erosion and, consequently, yield a longer delay as do coatings which are designed to dissolve above pH 7. Preferred coatings typically range from about 10 micron in thickness to about 3 mm in thickness and more preferably 10 um to 500 um.
When ingested, the twice-coated tablet passes through the stomach, where the second coating prevents release of the latrepirdine under the acidic conditions prevalent there. When the tablet passes out of the stomach and into the small intestine, where the pH is higher, the second coating erodes or dissolves according to the physicochemical properties of the chosen material. Upon erosion or dissolution of the second coating, the first coating prevents immediate or rapid release of the latrepirdine and modulates the release so as to prevent the production of high concentrations, thereby minimizing side-effects.
Another embodiment comprises a multiparticulate wherein each particle is dual coated as described above for tablets, first with a polymer designed to yield controlled release of the latrepirdine and then coated with a polymer designed to delay onset of release in the environment of the Gl tract when the dosage form is ingested. The beads contain latrepirdine and may contain one or more excipients as
needed for fabrication and performance. Multiparticulates which contain a high fraction of latrepirdine relative to binder are desired. The multiparticulate may be of a composition and be fabricated by any of the techniques previously disclosed for multiparticulates used to make reservoir systems (including extrusion and spheronization, wet granulation, fluid bed granulation, and rotary bed granulation, seed building, and so forth).
The controlled release coating may be as known in the art, especially polymer coatings, to fabricate the membrane, as previously discussed for reservoir systems. Suitable polymer coating materials, equipment, and coating methods also include those previously discussed.
The rate of latrepirdine release from the controlled-release-coated multiparticulates (e.g., the multiparticulates before they receive the delayed-release coating) and methods of modifying the coating are also controlled by the factors previously discussed for reservoir system latrepirdine multiparticulates.
The second membrane or coating for dual coated multiparticulates is a delayed-release coating which is applied over the first controlled-release coating, as disclosed above for tablets, and may be formed from the same materials. It should be noted that the use of the so-called "enteric" materials to practice this embodiment differs significantly from their use to produce conventional enteric dosage forms. With conventional enteric forms, the object is to delay release of the drug until the dosage form has passed the stomach and then to deliver the dose shortly after emptying from the stomach. Dosing of latrepirdine directly and completely to the duodenum is undesirable, however, due to local metabolism which is sought to be minimized or avoided by this invention. Therefore, if conventional enteric polymers are to be used to practice this embodiment, it may be necessary to apply them significantly more thickly than in conventional practice, in order to delay drug release until the dosage form reaches the lower Gl tract. However, it is preferred to effect a controlled delivery of latrepirdine after the delayed-release coating has dissolved or eroded, therefore the benefits of this embodiment may be realized with a proper combination of delayed-release character with controlled-release character, and the delayed-release part alone may or may not necessarily conform to USP enteric criteria. The thickness of the delayed-release coating is adjusted to give the desired delay property. In
general, thicker coatings are more resistant to erosion and, consequently, yield a longer delay.
It should also be noted, that sustained release osmotic systems as defined above, could also be defined in the current delay then controlled release category. Typical osmotic sustained release systems have an initial delay of 0.5-6 hours prior to drug release in a controlled fashion. In this manner, a standard osmotic monolithic or bilayer sustained release system embodies the definition of delay followed by controlled release.
Delayed Release and Immediate Release Components
A first delayed release embodiment according to the invention is a "pH- dependent coated tablet", which comprises a tablet core comprising latrepirdine, a disintegrant, a lubricant, and one or more pharmaceutical carriers, such core being coated with a material, preferably a polymer, which is substantially insoluble and impermeable at the pH of the stomach, and which is more soluble and permeable at the pH of the small intestine. In another embodiment, the coating polymer is substantially insoluble and impermeable at pH 5.0. It is preferred that the tablet core be coated with an amount of polymer sufficient to assure that substantially no release of latrepirdine from the dosage form occurs until the dosage form has exited the stomach and has resided in the small intestine for about 15 minutes or greater, preferably about 30 minutes or greater, thus assuring that minimal latrepirdine is released in the duodenum. Mixtures of a pH-sensitive polymer with a water-insoluble polymer may also be employed. Tablets are coated with an amount of polymer at a coating coverage from about 2 to 8 mg/cm2.
pH-sensitive polymers which are relatively insoluble and impermeable at the pH of the stomach, but which are more soluble and permeable at the pH of the small intestine and colon include polyacrylamides, phthalate derivatives such as acid phthalates of carbohydrates, amylose acetate phthalate, cellulose acetate phthalate, other cellulose ester phthalates, cellulose ether phthalates, hydroxypropylcellulose phthalate, hydroxypropylethylcellulose phthalate, hydroxypropylmethylcellulose phthalate, methylcellulose phthalate, polyvinyl acetate phthalate, polyvinyl acetate hydrogen phthalate, sodium cellulose acetate phthalate, starch acid phthalate, styrene-maleic acid dibutyl phthalate copolymer, styrene-maleic acid polyvinylacetate
phthalate copolymer, styrene and maleic acid copolymers, polyacrylic acid derivatives such as acrylic acid and acrylic ester copolymers, polymethacrylic acid and esters thereof, poly acrylic methacrylic acid copolymers, shellac, and vinyl acetate and crotonic acid copolymers.
Some preferred pH-sensitive polymers include shellac; phthalate derivatives, particularly cellulose acetate phthalate, polyvinylacetate phthalate, and hydroxypropylmethylcellulose phthalate; polyacrylic acid derivatives, particularly polymethyl methacrylate blended with acrylic acid and acrylic ester copolymers; and vinyl acetate and crotonic acid copolymers.
Cellulose acetate phthalate (CAP) may be applied to latrepirdine tablets to provide delayed release of latrepirdine until the latrepirdine-containing tablet has passed the sensitive duodenal region, that is to delay the release of latrepirdine in the gastrointestinal tract until about 15 minutes, and preferably about 30 minutes, after the latrepirdine-containing tablet has passed from the stomach to the duodenum. The CAP coating solution may also contain one or more plasticizers, such as diethyl phthalate, polyethyleneglycol-400, triacetin, triacetin citrate, propylene glycol, and others as known in the art. Some plasticizers are diethyl phthalate and triacetin. The CAP coating formulation may also contain one or more emulsifiers, such as polysorbate-80.
Anionic acrylic copolymers of methacrylic acid and methylmethacrylate are also particularly useful coating materials for delaying the release of latrepirdine from latrepirdine-containing tablets until the tablets have moved to a position in the small intestine which is distal to the duodenum. Copolymers of this type are available from RohmPharma Corp, under the trade names Eudragit-L.RTM. and Eudragit-S.RTM, Eudragit-FS. Eudragit-L.RTM., Eudragit-S.RTM. and Eudragit FS are anionic copolymers of methacrylic acid and methylmethacrylate. The ratio of free carboxyl groups to the esters is approximately 1 :1 in Eudragit-L.RTM. and approximately 1 :2 in Eudragit-S.RTM.. Mixtures of the Eudragits may also be used. For coating of latrepirdine-containing tablets, these acrylic coating polymers may be dissolved in an organic solvent or mixture of organic solvents or may be applied as aqueous colloidal suspensions. Useful solvents for the organic coating purpose are acetone, isopropyl alcohol, and methylene chloride. It is generally advisable to include 5-20% plasticizer
in coating formulations of acrylic copolymers. Useful plasticizers are polyethylene glycols, propylene glycols, diethyl phthalate, dibutyl phthalate, castor oil, and triacetin.
The delay time before release of latrepirdine, after the "pH-dependent coated tablet" dosage form has exited the stomach, may be controlled by the choice and the relative amounts of the Eudragits used in the coating. Eudragit-L.RTM. films dissolve above pH 6.0, and Eudragit-S.RTM. films dissolve above 7.0, Eudragit FS dissolves above 7.0, and mixtures dissolve at intermediate pH values. Since the pH of the duodenum is approximately 6.0 and the pH of the colon is approximately 7.0, coatings composed of mixtures of Eudragit-L.RTM. and Eudragit-S.RTM. provide protection of the duodenum from latrepirdine. If it is desired to delay release of latrepirdine until the latrepirdine-containing "pH-dependent coated tablet" has reached the colon, Eudragit-S.RTM. or Eudragit FS may be used as the coating material, as described by Dew et al (Br. J. Clin. Pharmac. 14 (1982) 405-408) and Cole et al (Int. J. Pharm., 231 (2002) 83-95). In order to delay the release of latrepirdine for about 15 minutes or more, preferably 30 minutes or more, after the dosage form has exited the stomach, preferred coatings comprise from about 9:1 to about 1 :9 Eudragit-L.RTM./Eudragit-S.RTM., more preferably from about 9:1 to about 1 :4 Eudragit-L.RTM./Eudragit-S.RTM.. The coating may comprise from about 3% to about 70% of the weight of the uncoated tablet core. Preferably, the coating comprises from about 5% to about 50% of the weight of the tablet core.
In a further embodiment, a "pH-dependent coated bead", beads (about 0.5 to 3.0 mm in diameter) comprising latrepirdine plus carrier are coated with one or more of the aforementioned pH-sensitive polymers. The coated beads may be placed in a capsule or may be compressed into a tablet, with care taken to avoid damaging the polymeric coat on individual beads during tablet compression. Preferred coated beads are those which exhibit substantially no release of latrepirdine from the dosage form until the beads have exited the stomach and have resided in the small intestine for about 15 minutes or greater, preferably about 30 minutes or greater, thus assuring that minimal latrepirdine is released in the duodenum. Mixtures of a pH- sensitive polymer with a water-insoluble polymer are also included. As described above, latrepirdine-containing beads may be coated with mixtures of polymers whose
solubilities vary at different pH's. For example, preferred coatings comprise from about 9:1 to about 1 :9 Eudragit-L.RTM./Eudragit-S.RTM., more preferably from 9:1 to 1 :4 Eudragit-L.RTM./Eudragit-S.RTM.. The coating may comprise from about 5% to about 200% of the weight of the uncoated bead core. Preferably, the coating comprises from about 10% to about 100% of the weight of the bead core.
In a further embodiment, ("pH-dependent coated particle"), small latrepirdine- containing particles (about 0.01 to 0.5 mm in diameter, preferably 0.05 to 0.5 mm in diameter) are coated with one or more of the aforementioned pH-sensitive polymers. The coated particles may be placed in a capsule or may be compressed into a tablet, with care taken to avoid damaging the polymeric coat on individual particles during tablet compression. Preferred coated particles are those which exhibit substantially no release of latrepirdine from the dosage form until the particles have exited the stomach and have resided in the small intestine for about 30 minutes or greater, preferably 60 minutes or greater more preferably 90 minutes or greater, thus assuring that minimal latrepirdine is released in the duodenum and upper part of the small intestine. Mixtures of a pH-sensitive polymer with a water-insoluble polymer are also included. Preferred latrepirdine-containing particles are coated with an amount of polymer comprising about 25% to about 200% of the weight of the uncoated latrepirdine-containing particle core.
A further embodiment constitutes a modification of the pH-dependent coated tablet, pH-dependent coated bead, and pH-dependent coated particle embodiments. The latrepirdine-containing core tablet, bead, or particle is first coated with a barrier coat, and then is coated with the pH-dependent coat. The function of the barrier coat is to separate latrepirdine from the pH-dependent coat. Suitable barrier coatings are composed of water-soluble materials such as sugars such as sucrose, or water- soluble polymers such as hydroxypropyl cellulose, hydroxypropyl methylcellulose, and the like. Hydroxypropyl cellulose and hydroxypropylmethylcellulose are preferred. The barrier coat may comprise from about 1 % to about 15%, preferably from about 2% to about 10%, of the weight of the uncoated latrepirdine-containing tablet, bead or particle core.
Coating of latrepirdine-containing tablets, beads and particles may be carried out using equipment known in the art. For example, latrepirdine-containing tablet
cores may be coated with a pan-coater, such as a Hi-Coater (Freund Corp.), or an Accela-Cota (Manesty Corp., Liverpool). Latrepirdine-containing beads and particles are preferably coated using a fluidized bed coater, such as a Wurster coater, utilizing coating equipment available for example from the Glatt Corporation (Ramsey, N.J.). Beads may also be coated using a rotary granulator, such as a CF-granulator available from Freund Corp.
Bursting Osmotic Beads and Cores (Pulsatile Delivery)
In a further embodiment ("bursting osmotic core device"), latrepirdine is incorporated in an osmotic bursting device which comprises a tablet core or bead core containing latrepirdine and, optionally, one or more osmagents. Devices of this type have been generally disclosed in Baker, U.S. Pat. No. 3,952,741 , which is incorporated herein by reference. Examples of osmagents are sugars such as glucose, sucrose, mannitol, lactose, and the like; and salts such as sodium chloride, potassium chloride, sodium carbonate, and the like; water-soluble acids such as tartaric acid, fumaric acid, and the like. The latrepirdine-containing tablet core or bead core is coated with a polymer which forms a semipermeable membrane, that is, a membrane which is permeable to water but is substantially impermeable to latrepirdine. Examples of polymers which provide a semipermeable membrane are cellulose acetate, cellulose acetate butyrate, and ethylcellulose, preferably cellulose acetate. The semipermeable coating membrane may alternatively be composed of one or more waxes, such as insect and animal waxes such as beeswax, and vegetable waxes such as carnauba wax and hydrogenated vegetable oils. A melt mixture of a polyethylene glycol, e.g., polyethylene glycol-6000, and a hydrogenated oil, e.g., hydrogenated castor oil, may be used as a coating, as described for isoniazid tablets by Yoshino (Capsugel Symposia Series; Current Status on Targeted Drug Delivery to the Gastrointestinal Tract; 1993; pp.185-190). Some preferred semipermeable coating materials are cellulose esters and cellulose ethers, polyacrylic acid derivatives such as polyacrylates and polyacrylate esters, and polyvinyl alcohols and polyalkenes such as ethylene vinyl alcohol copolymer. Other semipermeable coating materials are cellulose acetate and cellulose acetate butyrate.
When a coated tablet or bead of the "bursting osmotic core" embodiment of this invention is placed in an aqueous environment of use, water passes through the semipermeable membrane into the core, dissolving a portion of the latrepirdine and osmagent, generating a colloidal osmotic pressure which results in bursting of the semipermeable membrane and release of latrepirdine into the aqueous environment. By choice of bead or tablet core size and geometry, identity and quantity of osmagent, and thickness of the semipermeable membrane, the time lag between placement of the dosage form into the aqueous environment of use and release of the enclosed latrepirdine may be chosen. It will be appreciated by those skilled in the art that increasing the surface-to-volume ratio of the dosage form, and increasing the osmotic activity of the osmagent serve to decrease the time lag, whereas increasing the thickness of the coating will increase the time lag. Osmotic-bursting devices of this invention are those which exhibit substantially no release of latrepirdine from the dosage form until the dosage form has exited the stomach and has resided in the small intestine for about 15 minutes or greater. Some osmotic-bursting devices exhibit substantially no release of latrepirdine from the dosage form until the dosage form has exited the stomach and has resided in the small intestine for about 30 minutes or greater. Other osmotic-bursting devices exhibit substantially no release of latrepirdine from the dosage form until the dosage form has exited the stomach and has resided in the small intestine for about 90 minutes or greater. Still other osmotic- bursting devices exhibit substantially no release of latrepirdine from the dosage form until the dosage form has exited the stomach and has resided in the small intestine for and most preferably 3 hours or greater, thus assuring that minimal latrepirdine is released in the duodenum and upper small intestine. A bursting osmotic core tablet or bead has a tablet or bead core which may contain from about 10-95% latrepirdine, about 0-60% osmagent, as described above, and about 5-20% other pharmaceutical aids such as binders and lubricants. The semipermeable membrane coating on a tablet, such as a cellulose acetate coating, is present at a weight corresponding to from about 2% to about 30%, preferably from about 3% to about 10%, of the weight of the tablet core. The semipermeable membrane coating on a bead, such as a cellulose acetate coating, is present at a weight corresponding to from about 2% to about 80% of the weight of the bead core. In another embodiment, the
semipermeable coating on a bead is present at a weight corresponding to from 3% to 30% of the weight of the bead core.
A bursting osmotic core device possesses no mechanism for "sensing" that the device has exited the stomach and entered the duodenum. Thus devices of this type release latrepirdine at a predetermined time after entering an aqueous environment, e.g., after being swallowed. In the fasted state, indigestible non- disintegrating solids, such as the "bursting osmotic core devices" of this invention, are emptied from the stomach during phase III of the Interdigestive Migrating Myoelectric Complex (IMMC), which occurs approximately every 2 hr in the human. Depending on the stage of the IMMC at the time of dosing in the fasted state, a bursting osmotic core device may exit the stomach almost immediately after dosing, or as long as 2 hr after dosing. In the fed state, indigestible non-disintegrating solids, which are <1 1 mm in diameter, will empty slowly from the stomach with the contents of the meal (Khosla and Davis, Int. J. Pharmaceut. 62 (1990) R9-R1 1 ). If the indigestible non-disintegrating solid is greater than about 1 1 mm in diameter, e.g., about the size of a typical tablet, it will be retained in the stomach for the duration of the digestion of the meal, and will exit into the duodenum during phase III of an IMMC, after the entire meal has been digested and has exited the stomach. The release of latrepirdine can be delayed until about 15 min or more. The release of latrepirdine can be delayed until 30 minutes or more. The release of latrepirdine can be delayed until about 90 minutes or greater. The release of latrepirdine can be delayed until about 3 hours or greater after the dosage form has exited the stomach. A bursting osmotic core device which releases latrepirdine about 3 hr or more after ingestion will show a decrease in metabolism as measured by a reduction of metabolic markers such as Amet. A bursting osmotic core device starts to release latrepirdine at about 2.5 hr after entering an aqueous environment, e.g., after ingestion, to more reliably assure that the device releases its latrepirdine distal to the duodenum, when dosed in the fasted state. Another "bursting osmotic core device" will start to release latrepirdine at about 4 hr after entering an aqueous environment. This 4 hr delay permits dosing in the fed state, and allows for an about 3.5 hr retention in the fed stomach, followed by an approximately 30 minute delay after the
dosage form has exited from the stomach. In this way, the release of latrepirdine into the most sensitive portion of the gastrointestinal tract, the duodenum, is minimized.
In a further embodiment, a "bursting coated swelling core", a latrepirdine- containing tablet or bead is prepared which also comprises 25-70% of a swellable material, such as a swellable colloid (e.g., gelatin), as described in Milosovich, U.S. Pat. No. 3,247,066, incorporated herein by reference. Swelling core materials are hydrogels, e.g., hydrophilic polymers which take up water and swell, such as polyethylene oxides, polyacrylic acid derivatives such as polymethyl methacrylate, polyacrylamides, polyvinyl alcohol, poly-N-vinyl-2-pyrrolidone, carboxymethylcellulose, starches, and the like. Swelling hydrogels for this embodiment include polyethylene oxides, carboxymethylcellulose and croscarmellose sodium. The colloid/hydrogel-containing latrepirdine-containing core tablet or bead is coated, at least in part, by a semipermeable membrane. Examples of polymers which provide a semipermeable membrane are cellulose acetate and cellulose acetate butyrate, and ethylcellulose. The semipermeable coating membrane may alternatively be composed of one or more waxes, such as insect and animal waxes such as beeswax, and vegetable waxes such as carnauba wax and hydrogenated vegetable oils. A melt mixture of a polyethylene glycol, e.g., polyethylene glycol-6000, and a hydrogenated oil, e.g., hydrogenated castor oil, may be used as a coating, as described for isoniazid tablets by Yoshino (Capsugel Symposia Series; Current Status on Targeted Drug Delivery to the Gastrointestinal Tract; 1993; pp.185-190). Some semipermeable coating materials are cellulose esters and cellulose ethers, polyacrylic acid derivatives such as polyacrylates and polyacrylate esters, polyvinyl alcohols and polyalkenes such as ethylene vinyl alcohol copolymer, cellulose acetate and cellulose acetate butyrate.
When a coated tablet or bead having a bursting coated swelling core is placed in an aqueous environment of use, water passes through the semipermeable membrane into the core, swelling the core and resulting in bursting of the semipermeable membrane and release of latrepirdine into the aqueous environment. By choice of bead or tablet core size and geometry, identity and quantity of swelling agent, and thickness of the semipermeable membrane, the time lag between placement of the dosage form into the aqueous environment of use and release of
the enclosed latrepirdine may be chosen. Preferred bursting coated swelling core devices of this invention are those which exhibit substantially no release of latrepirdine from the dosage form until the dosage form has exited the stomach and has resided in the small intestine for about 15 minutes or greater, preferably about 30 minutes or greater, thus assuring that minimal latrepirdine is released in the duodenum.
A bursting coated swelling core tablet or bead has a tablet or bead core which may contain from about 10-70% latrepirdine; about 15-60% swelling material, e.g., hydrogel; about 0-15% optional osmagent; and about 5-20% other pharmaceutical aids such as binders and lubricants. The semipermeable membrane coating on a tablet, preferably a cellulose acetate coating, is present at a weight corresponding to from about 2% to about 30%, preferably from 3% to 10%, of the weight of the tablet core. The semipermeable membrane coating on a bead, preferably a cellulose acetate coating, is present at a weight corresponding to from about 2% to about 80%, preferably from 3% to 30%, of the weight of the bead core.
A bursting coated swelling core device possesses no mechanism for sensing that the device has exited the stomach and entered the duodenum. Thus devices of this type release their latrepirdine contents at a predetermined time after entering an aqueous environment, e.g., after being swallowed, as previously discussed for bursting osmotic core devices, and the same consideration and preferences apply to making bursting coated swelling core devices.
In a further embodiment, a "pH-triggered osmotic bursting device", latrepirdine is incorporated into a device of the type described in allowed commonly assigned co-pending U.S. Pat. No. 5,358,502, issued Oct. 25, 1994, incorporated herein by reference. The device comprises latrepirdine and optionally one or more osmagents, surrounded at least in part by a semipermeable membrane. The semipermeable membrane is permeable to water and substantially impermeable to latrepirdine and osmagent. Useful osmagents are the same as those described above for bursting osmotic core devices. Useful semipermeable membrane materials are the same as those described above for bursting osmotic core devices. A pH- trigger means is attached to the semipermeable membrane. The pH-trigger means is activated by a pH above 5.0, and triggers the sudden delivery of the latrepirdine. In
this embodiment, the pH-trigger means comprises a membrane or polymer coating which surrounds the semipermeable coating. The pH-trigger coating contains a polymer which is substantially impermeable and insoluble in the pH range of the stomach, but becomes permeable and soluble at about the pH of the duodenum, about pH 6.0.
Exemplary pH-sensitive polymers are polyacrylamides, phthalate derivatives such as acid phthalates of carbohydrates, amylose acetate phthalate, cellulose acetate phthalate, other cellulose ester phthalates, cellulose ether phthalates, hydroxypropylcellulose phthalate, hydroxypropylethylcellulose phthalate, hydroxypropylmethylcellulose phthalate, methylcellulose phthalate, polyvinyl acetate phthalate, polyvinyl acetate hydrogen phthalate, sodium cellulose acetate phthalate, starch acid phthalate, styrene-maleic acid dibutyl phthalate copolymer, styrene- maleic acid polyvinylacetate phthalate copolymer, styrene and maleic acid copolymers, polyacrylic acid derivatives such as acrylic acid and acrylic ester copolymers, polymethacrylic acid and esters thereof, poly acrylic methacrylic acid copolymers, shellac, and vinyl acetate and crotonic acid copolymers.
Preferred pH-sensitive polymers include shellac; phthalate derivatives, particularly cellulose acetate phthalate, polyvinylacetate phthalate, and hydroxypropylmethylcellulose phthalate; polyacrylic acid derivatives, particularly polymethyl methacrylate blended with acrylic acid and acrylic ester copolymers; and vinyl acetate and crotonic acid copolymers. As described above cellulose acetate phthalate is available as a latex under the tradename Aquateric.RTM. (registered trademark of FMC Corp., Philadelphia, Pa.), and acrylic copolymers are available under the tradenames Eudragit-R.RTM. and Eudragit-L.RTM.. For appropriate application in this embodiment, these polymers should be plasticized utilizing plasticizers described above. The pH-trigger coating may also comprise a mixture of polymers, for example cellulose acetate and cellulose acetate phthalate. Another suitable mixture comprises Eudragit-L.RTM. and Eudragit-S.RTM.; the ratio of the two, and the coating thickness, defining the sensitivity of the "trigger", e.g., the pH at which the outer pH-trigger coating weakens or dissolves.
A pH-triggered osmotic bursting device generally operates as follows. After oral ingestion, the pH-trigger coating, which surrounds the semipermeable coating,
which in turn surrounds the latrepirdine-containing core tablet or bead, remains undissolved and intact in the stomach. In the stomach, water may or may not commence penetration through the pH-trigger coating and the semipermeable coating, thus starting hydration of the core, which contains latrepirdine and optional osmagent. After the device has exited the stomach and has entered the small intestine, the pH-trigger coating rapidly disintegrates and dissolves, and water passes through the semipermeable coating, dissolving latrepirdine and optional osmagent within the core. As the colloidal osmotic pressure across the semipermeable coating exceeds some threshold value, the semipermeable coating fails, and the device bursts, releasing latrepirdine. It is preferred that this bursting and release of latrepirdine occur at about 15 minutes or more, preferably 30 minutes or more, after the pH-triggered osmotic bursting device exits the stomach and enters the duodenum, thus minimizing exposure of the sensitive duodenum to latrepirdine.
For a pH-triggered osmotic bursting device, the lag-time or delay-time is controlled by the choice and amount of osmagent in the core, by the choice of semipermeable coating, and by the thickness of the semipermeable coating. It will be appreciated by those skilled in the art, for example, that a thicker semipermeable coating will result in a longer delay after the device has exited the stomach. A preferred pH-triggered osmotic bursting device is a bead or tablet core of latrepirdine with optional osmagent, coated with a 3-20% by weight cellulose acetate membrane, coated with a 3-20% by weight membrane composed of about 1 : 1 cellulose ace tat e/cellu lose acetate phthalate. Another preferred pH-triggered osmotic bursting device is a bead or tablet core of latrepirdine with optional osmagent, coated with a 3-20% by weight cellulose acetate membrane, coated with a 3-20% by weight membrane comprising from about 9:1 to about 1 : 1 Eudragit-L.RTM./Eudragit-S.RTM.
Advantageously, because a pH-triggered osmotic bursting device possesses a mechanism for sensing that the device has exited the stomach, intersubject variability in gastric emptying is not significant.
In a further embodiment, a "pH-triggered bursting coated swelling core", a tablet core or bead containing latrepirdine and a swelling material is coated with a semipermeable coating which is further coated with a pH-sensitive coating. The core composition, including choice of swelling material is as described above for the
bursting coated swelling core embodiment. The choice of semipermeable coating material and pH-sensitive coating material are as described above for the "pH- triggered osmotic core" embodiment. This device is described in detail in commonly- assigned copending U.S. patent application Ser. No. 08/023,227, filed Feb. 25, 1993, incorporated herein by reference.
A pH-triggered bursting swelling core embodiment generally operates as follows. After oral ingestion, the pH-trigger coating, which surrounds the semipermeable coating, which in turn surrounds the latrepirdine-containing core tablet or bead, remains undissolved and intact in the stomach. In the stomach, water may or may not commence penetration through the pH-trigger coating and the semipermeable coating, thus starting hydration of the core, which contains latrepirdine and water-swellable material, preferably a hydrogel. When the pH- triggered bursting swelling core device exits the stomach and enters the small intestine, the pH-trigger coating rapidly disintegrates and dissolves, and water passes through the semipermeable coating, dissolving latrepirdine and swelling the water-swellable material within the core. As the swelling pressure across the semipermeable coating exceeds some threshold value, the semipermeable coating fails, and the device bursts, releasing latrepirdine. This bursting and release of latrepirdine occurs at about 15 minutes or more, around about 30 minutes, after the pH-triggered bursting swelling core device exits the stomach and enters the duodenum, thus minimizing exposure of the sensitive duodenum to latrepirdine.
For the "pH-triggered bursting swelling core" device, the lag-time or delay- time can be controlled by the choice and amount of swelling material in the core, by the choice of semipermeable coating, and by the thickness of the semipermeable coating. It will be appreciated by those skilled in the art, for example, that a thicker semipermeable coating will result in a longer delay after the device has exited the stomach. A pH-triggered bursting swelling core device contains a bead or tablet core of latrepirdine with synthetic hydrogel, preferably carboxymethylcellulose, coated with a 3-20% by weight cellulose acetate membrane, coated with a 3-20% by weight membrane composed of about 1 : 1 cellulose acetate/cellulose acetate phthalate. Another pH-triggered bursting swelling core device contains a bead or tablet core of latrepirdine with synthetic hydrogel, preferably carboxymethylcellulose, coated with a
3-20% by weight cellulose acetate membrane, coated with a 3-20% by weight membrane composed of from about 9: 1 to about 1 :1 Eudragit-L.RTM./Eudragit- S.RTM.
Advantageously, because a pH-triggered bursting swelling core device possesses a mechanism for sensing that the device has exited the stomach, intersubject variability in gastric emptying is not significant.
A current review of this bursting technology is Journal of Controlled Release; 134 (2009) 74-80 and herein incorporated as reference in its entirety.
Delayed release embodiments of the invention are solid dosage forms for oral administration comprising latrepirdine and a pharmaceutically acceptable carrier, which release not more than 10% of their incorporated latrepirdine into a mammal's stomach, and which release not more than an additional 10% during the first 15 minutes after entering said mammal's duodenum. The timing of release of latrepirdine in the stomach or duodenum may be tested utilizing a variety of approaches including, but not limited to, x-ray evaluation, nuclear magnetic resonance imaging, gamma scintigraphy, or direct sampling of the gastric and duodenal contents via intubation. These tests, while possible, can be very difficult to carry out in humans. A more convenient test for a delayed release embodiment of the current invention is a two stage in vitro dissolution test which was previously described above as "in vitro dissolution test 2".
BRIEF DESCRIPTION OF THE DRAWINGS
The invention is further described by the following nonlimiting examples, which refer to the accompanying Figures 1 through 4, short particulars of which are given below.
Figure 1 : Representative in vitro dissolution profiles for latrepirdine immediate release tablets measured using dissolution test 1 .
Figure 2: Representative in vitro dissolution profiles for 90 and 200 wt% enteric coated Eudragit FS30 melt spray congeal multiparticulates. Dissolution was measured using dissolution test 2.
Figure 3: Representative in vitro dissolution profile 200 wt% enteric coated Eudragit L30D-55 melt spray congeal multiparticulates. Dissolution was measured using dissolution test 2.
Figure 4: Representative in vitro dissolution profiles for 10.5 wt% and 21 wt% AMT 3: 1 CA:HPC membrane coatings. Dissolution was completed by dissolution test 1.
EXAMPLES
Preparation A - Latrepirdine Immediate Release Core Tablets

Footnotes:
a Note the dose and strength of the tablets is defined in terms of weight of latrepirdine dihydrochloride (a 5mg tablet is 4.1mg of latrepirdine free base; (a 10 mg tablet is 8.2 mg of latrepirdine free base); (a 20 mg tablet is 16.4 mg of Latrepirdine free base) . The API has a latrepirdine dihydrochloride theoretical factor of 91.59% (to correct for the dihydrate).
b weight adjusted for potency of latrepirdine dihydrochloride. Avicel PH102, FMC Corporation c Fast Flo, Foremost Farms
d Glycolys, Roquette
8 Vegetable sourced; Mallinckrodt, added intragranularly
' Vegetable sourced; Mallinckrodt, added extragranularly
9 Opadry II Green (85F 11805) for 5 mg tablet; Opadry II White (Y-30-18037) for 10 mg. tablet;
Opadry II Pink (32K 14828)for 20 mg. tablet
h Removed (evaporated) during film coating/drying and does not appear in the final product. Manufacturing Process For the preparation of the 5, 10 and 20 mg latrepirdine tablets
The excipients from the above table (microcrystalline cellulose, lactose and sodium starch glycolate) and latrepirdine di-HCI were loaded into an appropriately sized bin, ensuring that the latrepirdine di-HCI is sandwiched between the excipients. The premix was blended for 10 minutes at 12 +/- 1 RPM and then passed through a rotary mill equipped with a 1 .0mm screen, running at approximately 1000 RPM.
The sieved blend was collect in an appropriately sized bin and to this was added intragranular magnesium stearate and blend for 5 minutes at 12 +/- 1 RPM.
The lubricated blend was processed through a Gerteis roller compactor equipped with an inline oscillated mill and the granules were collected into an appropriately sized bin. The targeted ribbon solid fraction was 0.67 (0.65-0.72). The collected granules were blended in an appropriately sized bin. To this was added extragranular magnesium stearate and this mixture was blended for 5 minutes at 12 +/- 1 RPM.
Tablets were compressed on a rotary tablet press to an average target weight of l OO.Omg, a target hardness of 5-9 kP, and a target thickness of 2.4-2.9 mm. The tablets were passed through a deduster. An aqueous film-coating was applied to the tablets to a target weight gain of 4.0% using a perforated coating pan.
EXAMPLE 1
Production of Melt-Spray-Conqeal Microsphere Cores for Latrepirdine
A 3 kg batch of microspheres was prepared as follows: 1230 g of glyceryl behenate was added to an 8 quart V-Blender and blended for 1 minute at 12 ± 1 RPM to coat the metal surfaces. 540 g of latrepirdine dihydrochloride dihydrate was then added to the center of the blender. Another 1230 g of glyceryl behenate was added to the blender ensuring that the latrepirdine was completely covered. The resulting mixture was blended for 15 minutes at 12 ± 1 RPM. The blended mixture
was then discharged into a plastic charge bag and transferred to a K-Tron Soder powder feeder. The powder feeder was calibrated to deliver the blend at a rate of 50 ± 5 g/min. The blend was processed through a Leistritz 17mm twin-screw extruder fed by the K-Tron Soder powder feeder and coupled to the melt-spray-congeal (MSC) spinning disk according to the operating conditions in Table 1. The particle size of the resulting microspheres was D[4,3] = 155 ± 10 micron measured by a Sympatec dynamic-light-scattering particle size analyzer with a measuring range of 9.0 to 1750 μιτι, a cycle time of 10 ms, a feed pressure of 2.0 bar, and a vacuum pressure of 74 mbar.
Table 1. Powder Feeder, Twin-Screw Extruder and Melt-Spray-Congeal
Operating Conditions
K-Tron Powder Feeder Value
Feed Rate 50 ± 5 g/min
Leistritz Twin-Screw Value
Extruder
Screw Speed 150 ± 5 rpm Barrel 1 (Feed Zone) 20 ± 5°C
Barrel 2 40 ± 5°C
Barrel 3 60 ± 5°C
Barrel 4 90 ± 5°C
Barrel 5 90 ± 5°C
Gate Adapter 90 ± 5°C
Die Adapter 90 ± 5°C
Melt-Spray-Congeal Value
Jacket Water Temperature 92 ± 3°C
MSC Disk Heater 91 ± 3°C
Temperature
Melt Temperature 90 ± 3°C
MSC Disk Speed 4250 ± 50 rpm
The microspheres were transferred to trays and annealed in a temperature and relative humidity controlled oven at 40°C/75% RH for 24 hours with a maximum bed depth of 0.5 inch. After annealing the microspheres were passed through a 425 micron screen to break up any weak agglomerates formed during annealing.
EXAMPLE 2
Preparation of 90 wt% Eudraqit FS30D Enteric Coated Latrepirdine
Microspheres
162.9 g of purified water was added to an appropriately sized container and heated on a hot plate while stirring until the water temperature reached 70-75°C. Meanwhile, 4 g of triethyl citrate and 4 g of polysorbate 80 were added to a separate container, and 3.2 g of glyceryl monostearate was weighted out onto a piece of wax paper and set aside. Once the water was heated to 70-75°C it was poured into the container with the triethyl citrate and polysorbate 80. The glyceryl monostearate was added to the water/triethyl citrate/polysorbate 80 mixture while homogenizing on a high-shear mixer at 3500-4000 rpm. The overall mixture was homogenized for 10-15 minutes. Another 162.9 g of purified water was added to the homogenized glyceryl monostearate suspension and was mixed with a conventional stirrer until the suspension cooled down below 30°C. 263.2 g of Eudragit® FS30D dispersion was weighed out into a separate container. The cooled glyceryl monostearate suspension was then poured slowly into the Eudragit® FS30D dispersion while mixing gently. The final coating solution was passed through a 425 micron screen just before use.
The coating was carried out on a Glatt GPCG-1 fluid bed coater with a 3.5" ID product chamber and a Wurster column insert charged with 50 g of core microspheres from Example 1. The Wurster column gap was set at 5mm. A spray rate of 1 .5 - 2.2 g/min was maintained during the run with a product temperature of 26-28°C at an air flow setting of 0.3 bar. The microspheres were sprayed until 300 g of coating solution was deposited, which corresponds to a weight gain of approximately 90%. The inlet air heater was then turned off and the microspheres were dried under fluidization for 25 minutes. The resulting microspheres were passed through a 425 micron screen and any coated material greater than 425 micron was discarded. The sieved microspheres were then blended with 2% Talc by weight in a Turbula bottle blender.
EXAMPLE 3
Preparation of 200 wt% Eudragit FS30D Enteric Coated Latrepirdine
Microspheres
181.3 g of purified water was added to an appropriately sized container and heated on a hot plate while stirring until the water temperature reached 70-75°C. Meanwhile, 4.4 g of triethyl citrate and 4.4 g of polysorbate 80 were added to a separate container, and 3.6 g of glyceryl monostearate was weighted out onto a piece of wax paper and set aside. Once the water was heated to 70-75°C it was poured into the container with the triethyl citrate and polysorbate 80. The glyceryl monostearate was added to the water/triethyl citrate/polysorbate 80 mixture while homogenizing on a high-shear mixer at 3500-4000 rpm. The overall mixture was homogenized for 10-15 minutes. Another 181.3 g of purified water was added to the homogenized glyceryl monostearate suspension and was mixed with a conventional stirrer until the suspension cooled down below 30°C. 293 g of Eudragit® FS30D dispersion was weighed out into a separate container. The cooled glyceryl monostearate suspension was then poured slowly into the Eudragit® FS30D dispersion while mixing gently. The final coating solution was passed through a 425 micron screen just before use.
The coating was carried out on a Glatt GPCG-1 fluid bed coater with a 3.5" ID product chamber and a Wurster column insert charged with 25 g of core microspheres from Example 1. The Wurster column gap was set at 5mm. A spray rate of 1 .5 - 2.2 g/min was maintained during the run with a product temperature of 26-28°C at an air flow setting of 0.3 bar. The microspheres were sprayed until 334 g of coating solution was deposited, which corresponds to a weight gain of approximately 200%. The inlet air heater was then turned off and the microspheres were dried under fluidization for 25 minutes. The resulting microspheres were passed through a 425 micron screen and any coated material greater than 425 micron was discarded. The sieved microspheres were then blended with 2% Talc by weight in a Turbula bottle blender.
EXAMPLE 4
Preparation of 90 wt% Eudragit L30D-55 Enteric Coated Latrepirdine
Microspheres
42.2 g of purified water was added to an appropriately sized container and heated on a hot plate while stirring until the water temperature reached 70-75°C. Meanwhile, 3.8 g of triethyl citrate and 0.8 g of polysorbate 80 were added to a
separate container, and 2.0 g of glyceryl monostearate was weighted out onto a piece of wax paper and set aside. Once the water was heated to 70-75°C it was poured into the container with the triethyl citrate and polysorbate 80. The glyceryl monostearate was added to the water/triethyl citrate/polysorbate 80 mixture while homogenizing on a high-shear mixer at 3500-4000 rpm. The overall mixture was homogenized for 10-15 minutes. Another 42.2 g of purified water was added to the homogenized glyceryl monostearate suspension and was mixed with a conventional stirrer until the suspension cooled down below 30°C. 129 g of Eudragit® L30D-55 dispersion was weighed out into a separate container. The cooled glyceryl monostearate suspension was then poured slowly into the Eudragit® L30D-55 dispersion while mixing gently. The final coating solution was passed through a 425 micron screen just before use.
The coating was carried out on a Glatt GPCG-1 fluid bed coater with a 3.5" ID product chamber and a Wurster column insert charged with 25 g of core microspheres from Example 1. The Wurster column gap was set at 5mm. A spray rate of 1 .2 - 1.6 g/min was maintained during the run with a product temperature of 30-32°C at an air flow setting of 0.3 bar. The microspheres were sprayed until 1 10 g of coating solution was deposited, which corresponds to a weight gain of approximately 90%. The inlet air heater was then turned off and the microspheres were dried under fluidization for 20 minutes. The resulting microspheres were passed through a 425 micron screen and any coated material greater than 425 micron was discarded.
EXAMPLE 5
Preparation of 200 wt% Eudragit L30D-55 Enteric Coated Latrepirdine
Microspheres
93.2 g of purified water was added to an appropriately sized container and heated on a hot plate while stirring until the water temperature reached 70-75°C. Meanwhile, 8.6 g of triethyl citrate and 1.8 g of polysorbate 80 were added to a separate container, and 4.2 g of glyceryl monostearate was weighted out onto a piece of wax paper and set aside. Once the water was heated to 70-75°C it was poured into the container with the triethyl citrate and polysorbate 80. The glyceryl monostearate was added to the water/triethyl citrate/polysorbate 80 mixture while
homogenizing on a high-shear mixer at 3500-4000 rpm. The overall mixture was homogenized for 10-15 minutes. Another 93.2 g of purified water was added to the homogenized glyceryl monostearate suspension and was mixed with a conventional stirrer until the suspension cooled down below 30°C. 285.2 g of Eudragit® L30D-55 dispersion was weighed out into a separate container. The cooled glyceryl monostearate suspension was then poured slowly into the Eudragit® L30D-55 dispersion while mixing gently. The final coating solution was passed through a 425 micron screen just before use.
The coating was carried out on a Glatt GPCG-1 fluid bed coater with a 3.5" ID product chamber and a Wurster column insert charged with 25 g of core microspheres from Example 1. The Wurster column gap was set at 5mm. A spray rate of 1 .3 - 1.8 g/min was maintained during the run with a product temperature of 30-32°C at an air flow setting of 0.3 bar. The microspheres were sprayed until 243 g of coating solution was deposited, which corresponds to a weight gain of approximately 200%. The inlet air heater was then turned off and the microspheres were dried under fluidization for 20 minutes. The resulting microspheres were passed through a 425 micron screen and any coated material greater than 425 micron was discarded.
EXAMPLE 6
Preparation of drug-layered core
A drug layer solution is prepared by adding 1246.67g purified water to a container equipped with a mixer. With vigorous mixing, 20. Og hydroxypropyl methylcellulose (Methocel E5), and 200. OOg latrepirdine hydrochloride are dissolved therein. Mixing is continued until complete dissolution is achieved.
Non-pareil seeds (1 OOOg, 20/25 mesh,) are dispensed into a Wurster fluid bed coater (Glatt 1 .1 ). After fluidization of the non-pareils, spraying of the drug layer solution is commenced to layer drug solution effectively onto the seeds. Spraying is continued until the drug layer solution is exhausted (approximately 22% w/w weight gain). The beads are then sieved through a 710μιτι screen and an 850μιτι to remove any agglomerated or broken beads.
An isolation layer solution is prepared by adding 664.0 g of water to a container equipped with a mixer. With vigorous mixing, 50. Og hydroxypropyl methylcellulose (methocel E5) is mixed until a complete dissolution is achieved.
The drug layer cores are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidization of the drug layered cores, spraying of the isolation layer is commenced to layer the isolation solution onto the cores. Spraying is continued until all the solution is exhausted (approximately a 5% w/w weight gain). The beads are dried under the same conditions for 5 minutes. The beads are then sieved through a 710μιτι screen and an 850μιτι screen to remove any agglomerated or broken beads.
The beads are to be transferred to trays and annealed in a temperature and relative humidity controlled oven at 40°C/75% RH for at least 24 hours.
EXAMPLE 7
Preparation of drug-layered core
A drug layer solution is prepared by adding 1246.67g purified water to a container equipped with a mixer. With vigorous mixing, 20. Og hydroxypropyl methylcellulose (Methocel E5), and 200. OOg latrepirdine hydrochloride are dissolved therein. Mixing is continued until complete dissolution is achieved.
Microcrystalline Cellulose (MCC) seeds (1000g, 20/25 mesh,) are dispensed into a Wurster fluid bed coater (Glatt 1 .1 ). After fluidization of the MCC seeds, spraying of the drug layer solution is commenced to layer drug solution effectively onto the seeds. Spraying is continued until the drug layer solution is exhausted (approximately 22% w/w weight gain). The beads are then sieved through a 710μιτι screen and an 850μιτι to remove any agglomerated or broken beads.
An isolation layer solution is prepared by adding 664.0 g of water to a container equipped with a mixer. With vigorous mixing, 50. Og hydroxypropyl methylcellulose (methocel E5) is mixed until a complete dissolution is achieved.
The drug layer cores are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidization of the drug layered cores, spraying of the isolation layer is commenced to layer the isolation solution onto the cores. Spraying is continued until all the solution is exhausted (approximately a 5% w/w weight gain). The beads are dried under the same conditions for 5 minutes. The beads are then sieved through a 710μιτι screen and an 850μιτι screen to remove any agglomerated or broken beads.
The beads are to be transferred to trays and annealed in a temperature and relative humidity controlled oven at 40°C/75% RH for at least 24 hours.
EXAMPLE 8
Latrepirdine Enteric Coated Beads
A controlled release layer is prepared by adding 60 g of water to a container equipped with a mixer and heating to 70-80°C With vigorous mixing, add 6.13g of triethyl citrate, 7.60 g of glyceryl monostearate and 7.60g of Polysorbate 80 (as 33% aqueous solution). Add 144.60g of water to a container equipped with a mixer and with mixing add the emulsion previously prepared with mixing and allow to cool to room temperature. Add this dispersion to 279.33g of Methacrylic Acid - Ethyl Acrylate Copolymer (1 : 1 ) Dispersion 30 Per Cent (Eudragit® L30-D55) in a suitable container with a mixer and stir gently. Prior to coating pour the coating suspension through a 0.5 mm screen to remove any large agglomerates.
The drug layer cores as described in example 1 or 2 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidization of the drug layered cores, spraying of the controlled release layer suspension is commenced to layer the controlled release suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 10% w/w weight gain). The beads are dried under the same conditions for 10 minutes. The beads are then sieved through a 850μιτι screen and an 1000μιτι screen to remove any agglomerated or broken beads.
EXAMPLE 9
Latrepirdine Enteric Coated Beads
A controlled release layer is prepared by adding 90 g of water to a container equipped with a mixer and heating to 70-80°C With vigorous mixing, add 10.67g of triethyl citrate, 13.33 g of glyceryl monostearate and 13.33g of Polysorbate 80 (as 33% aqueous solution). Add 268.07g of water to a container equipped with a mixer and with mixing add the emulsion previously prepared with mixing and allow to cool to room temperature. Add this dispersion to 488.87g of Methacrylic Acid - Ethyl Acrylate Copolymer (1 : 1 ) Dispersion 30 Per Cent (Eudragit® L30-D55) in a suitable container with a mixer and stir gently. Prior to coating pour the coating suspension through a 0.5 mm screen to remove any large agglomerates.
The drug layer cores as described in Example 6 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidization of the drug layered cores, spraying of the controlled release layer suspension is commenced to layer the controlled release suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 17.5% w/w weight gain). The beads are dried under the same conditions for 10 minutes. The beads are then sieved through a 850μιτι screen and an 1000μιτι screen to remove any agglomerated or broken beads.
EXAMPLE 10
Latrepirdine Enteric Coated Beads
A controlled release layer is prepared by adding 150 g of water to a container equipped with a mixer and heating to 70-80°C With vigorous mixing, add 15.27g of triethyl citrate, 19.0 g of glyceryl monostearate and 19.0g of Polysorbate 80 (as 33% aqueous solution). Add 361.53g of water to a container equipped with a mixer and with mixing add the emulsion previously prepared to Methacrylic Acid - Ethyl Acrylate Copolymer (1 : 1 ) Dispersion 30 Per Cent (Eudragit® L30-D55) in a suitable container with a mixer and stir gently. Prior to coating pour the coating suspension through a 0.5 mm screen to remove any large agglomerates.
The drug layer cores as described in Example 6 or 7 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidization of the drug layered cores, spraying of the controlled release layer is commenced to layer the controlled release suspension onto the cores. Spraying is continued until all the suspension is
exhausted (approximately a 25% w/w weight gain). The beads are dried under the same conditions for 10 minutes. The beads are then sieved through a 850μιτι screen and an 1000μιτι screen to remove any agglomerated or broken beads.
EXAMPLE 11
Latrepirdine Enteric Coated Beads
A controlled release layer is prepared by adding 60 g of water to a container equipped with a mixer and heating to 70-80°C With vigorous mixing, add 6.13g of triethyl citrate, 7.60 g of glyceryl monostearate and 7.60g of Polysorbate 80 (as 33% aqueous solution). Add 144.60g of water to a container equipped with a mixer and with mixing add the emulsion previously prepared with mixing and allow to cool to room temperature. Add this dispersion to 279.33g of Methacrylic Acid - Ethyl Acrylate Copolymer (1 : 1 ) Dispersion 30 Per Cent (Eudragit® FS30 D) in a suitable container with a mixer and stir gently. Prior to coating pour the coating suspension through a 0.5 mm screen to remove any large agglomerates.
The drug layer cores as described in Example 6 or 7 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidization of the drug layered cores, spraying of the controlled release layer suspension is commenced to layer the controlled release suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 10% w/w weight gain). The beads are dried under the same conditions for 10 minutes. The beads are then sieved through a 850μιτι screen and an 1000μιτι screen to remove any agglomerated or broken beads.
EXAMPLE 12
Latrepirdine Enteric Coated Beads
A controlled release layer is prepared by adding 90 g of water to a container equipped with a mixer and heating to 70-80°C With vigorous mixing, add 10.67g of triethyl citrate, 13.33 g of glyceryl monostearate and 13.33g of Polysorbate 80 (as 33% aqueous solution). Add 268.07g of water to a container equipped with a mixer and with mixing add the emulsion previously prepared with mixing and allow to cool to room temperature. Add this dispersion to 488.87g of Methacrylic Acid - Ethyl Acrylate Copolymer (1 :1 ) Dispersion 30 Per Cent (Eudragit® FS30 D) in a suitable container with a mixer and stir gently. Prior to coating pour the coating suspension through a 0.5 mm screen to remove any large agglomerates.
The drug layer cores as described in example 6 or 7 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidization of the drug layered cores, spraying of the controlled release layer suspension is commenced to layer the controlled release suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 17.5% w/w weight gain). The beads are dried under the same conditions for 10 minutes. The beads are then sieved through a 850μιτι screen and an 1000μιτι screen to remove any agglomerated or broken beads.
EXAMPLE 13
Latrepirdine Enteric Coated Beads
A controlled release layer is prepared by adding 150 g of water to a container equipped with a mixer and heating to 70-80°C With vigorous mixing, add 15.27g of triethyl citrate, 19.0 g of glyceryl monostearate and 19.0g of Polysorbate 80 (as 33% aqueous solution). Add 361.53g of water to a container equipped with a mixer and with mixing add the emulsion previously prepared to Methacrylic Acid - Ethyl Acrylate Copolymer (1 : 1 ) Dispersion 30 Per Cent (Eudragit® FS30 D) in a suitable container with a mixer and stir gently. Prior to coating pour the coating suspension through a 0.5 mm screen to remove any large agglomerates.
The drug layer cores as described in example 6 or 7 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidization of the drug layered cores, spraying of the controlled release layer is commenced to layer the controlled release suspension onto the cores. Spraying is continued until all the solution is exhausted (approximately a 25% w/w weight gain). The beads are dried under the same conditions for 10 minutes. The beads are then sieved through a 850μιτι screen and an 1000μιτι screen to remove any agglomerated or broken beads.
EXAMPLE 14
Latrepirdine Tablet Osmotic Cores
The metal surfaces of a 25L bin were pre-coated by adding the batch quantity (also see Table 2 below), 2199.20g, of Avicel (PH 102) and blending for 1 minute at 12 +/- 1 RPM. The Fumaric acid, 800g, and one-half the batch quantity of mannitol, 2198.8g, were added to a 25L sized plastic charge bag and gently mixed together by inverting the bag. The 523.2g of latrepirdine 2HCI, was added to the center of the bin, meanwhile saving the container it was weighed in*. The latrepirdine 2HCI container
was rinsed twice with portions of the remaining mannitol, which was subsequently added to the bin. The contents of the plastic charge bag were emptied into the bin, covering the latrepirdine 2HCI. The plastic charge bag was saved. All of the components were blended in the bin for 10 minutes at 12 +/- 1 RPM.
The blend was passed through a Comil rotary mill equipped with screen and a round edge impeller running at approximately 750 RPM. The blend was collected in the saved plastic charge bag. The bin and mill were flushed to remove any residual API by adding the remaining mannitol to the bin, blending for 1 minute at 12 +/- 1 RPM, then releasing contents through the mill into the plastic charge bag. The milled blend was then transferred back to the bin blender. The plastic charge bag was saved. The bin contents were blended for 10 minutes at 12 +/- 1 RPM.
Intragranular magnesium stearate, 40g**, was added to the bin and blended for 5 minutes at 12 +/- 1 RPM. The lubricated blend was processed through a Gerteis roller compactor equipped with knurled rollers, side rims, and an inline oscillating mill containing a pocket rotor and a 0.8mm rasping plate. The target ribbon solid fraction was 0.71 (0.70-0.75) and granules were collected in the saved plastic charge bag.
The collected granules were transferred back to the bin blender and blended for 10 minutes at 12 +/- 1 RPM. Extragranular magnesium stearate, 40g**, was added to the bin and contents were blended for 5 minutes at 12 +/- 1 RPM. Final blend was affixed above a Kilian T-100 rotary tablet press. Tablets were compressed using 5/16" SRC tooling, to an average target weight of 250. Omg +/- 3% and a target hardness of 7.5 kP. Tablets were passed through a deduster and a metal detector. * All processes here forth were conducted using a closed system approach. Transfers between the bin, bags, the mill, Gerteis and Tablets press were executed using valve connections.
** A yield calculation was performed. Intragranular and extra granular magnesium stearate level should be adjusted if yield is less than 98%.
Table 2. 15mqA Latrepirdine Tablet Core Formulation Components
Item # Component Wt % Grams Use
Latrepirdine 2HCI * 6.54 523.20 API
Microcrystalline
2 27.49 2199.20 Dliuent cellulose (PH 102)
Mannitol 2080,
3 54.97 4397.60 Osmogen gran.
4 Fumaric Acid 10.00 800.00 Acid
Magnesium
stearate 0.50 40.00 Lubricant
(intragranular)
Magnesium
stearate 0.50 40.00 Lubricant
(extragranular)
Total 100.00 8000.00
* Assumes 91.7% potency
8000.00
factor
(calculating as the free base plus dihyrochloride minus
dihydrate)
EXAMPLE 15
Small Scale (~1 kq) Osmotic CR Asymmetric Membrane Technology (AMT) Short Duration Tablet Coating Process on Latrepirdine 15mq A Tablet Cores
Latrepirdine di-hydrochloride, 250mg (15mgA) tablet cores were coated by a functional solvent based film coating spraying process using a Vector LDCS-5 pan coater at small scale (1 kg) with an osmotic polymeric film coating with cellulose acetate as the polymer and hydroxypropyl cellulose as the plasticizer. The acetone and water are the co-solvents uses to dissolve the solids and facilitate pore formation. The coating solution composition contains 8% solids by weight, comprised of 6% cellulose acetate and 2% hydroxypropyl cellulose (3: 1 ratio) dissolved in 69% acetone and 23% water (3:1 ratio). The final dry target of 10.5 wt% coating will be -26 mg of dried coating (75% CA & 25% HPC) per tablet core (on a 250 mg core) to obtain 80% released in 8 hours by in vitro dissolution test 1 .
Table 3. Latrepirdine 15mgA tablets coated with a 10.5wt% short duration osmotic coating
Composition of short duration coated °o in mg tablet coat w/w tablet coating %)
Latrepirdine Tablet Core 250.0
Cellulose Acetate (Type 398-10) 6.0 19.7 7.875
Hydroxypropyl Cellulose (Klucel EF) 2.0 6.6 2.625
Acetone 69.0 (226.8)*
Purified water 23.0 (75.6)*
Total Weight 100% 276.3 10.50
* Volatile, not present in final dosage form
The AMT coating was prepared as a 8% (w/w) solution. The coating solution was applied to the tablet cores using a Vector LDCS-5 pan coater using the following parameters:
Table 4. Coating parameters used for AMT coating in the Vector LDCS-5 pan coater
Coating Parameter Target Value
Coater type Vector LDCS-5 pan coater
Coating pan 1.3L fully perforated with 4 baffles
Weight tablet cores (g) 1000
Pan RPM 15
Spray Rate (g/min/gun) 20
Number of spray guns 1
Gun-to-Bed distance 2.5
(inches)
Atomization Air (PSI) 20
Pattern Air (PSI) 10
Inlet Temperature (°C) 35
Outlet Temperature (°C) 23
Pan Air Flow (CFM) 40
Total solution sprayed (g) 1200
EXAMPLE 16
Small Scale (~1 kg) Osmotic CR Asymmetric Membrane Technology (AMT) Long Duration Tablet Coating Process on Latrepirdine 15mgA Tablet Cores
Latrepirdine di-hydrochloride, 250mg (15mgA) tablet cores were coated by a functional solvent based film coating spraying process using a Vector LDCS-5 pan coater at small scale (1 kg) with an osmotic polymeric film coating with cellulose acetate as the polymer and hydroxypropyl cellulose as the plasticizer. The acetone and water are the co-solvents uses to dissolve the solids and facilitate pore formation. The coating solution composition contains 8% solids by weight, comprised of 6% cellulose acetate and 2% hydroxypropyl cellulose (3: 1 ratio) dissolved in 69% acetone and 23% water (3:1 ratio). The final dry target of 21 wt%
coating will be -52.5 mg of dried coating (75% CA & 25% HPC) per tablet core (on a 250 mg core) to obtain 80% released in 12 hours by in vitro dissolution test 1.
Table 5. Latrepirdine 15mqA tablets coated with a 21wt% long duration osmotic coating

* Volatile, not present in final dosage form
The AMT coating was prepared as a 8% (w/w) solution. The coating solution was applied to the tablet cores using a Vector LDCS-5 pan coater using the following parameters: Table 6. Coating parameters used for AMT coating in the Vector LDCS-5 pan
coater
Coating Parameter Target Value
Coater type Vector LDCS-5 pan coater
Coating pan 1.3L fully perforated with 4 baffles
Weight tablet cores (g) 1000
Pan RPM 15
Spray Rate (g/min/gun) 20
Number of spray guns 1
Gun-to-Bed distance 2.5
(inches)
Atomization Air (PSI) 20
Pattern Air (PSI) 10
Inlet Temperature (°C) 35
Outlet Temperature (°C) 23
Pan Air Flow (CFM) 40
Total solution sprayed (g) 2500
EXAMPLE 17
Small Scale (~1 kq) Osmotic CR Semi-permeable membrane with a hole- Duration 1 (T«n=14h) Tablet Coating Process on Latrepirdine 15mqA Tablet
Cores
Latrepirdine di-hydrochloride, 250mg (15mgA) tablet cores were coated by a functional solvent based film coating spraying process using a Vector LDCS-5 pan coater at small scale (1 kg) with an osmotic polymeric film coating with cellulose acetate as the polymer and hydroxypropyl cellulose as the plasticizer. The acetone and water are the co-solvents uses to dissolve the solids and facilitate pore formation. The coating solution composition contains 5% solids by weight, comprised of 3.5% cellulose acetate and 1 .5% hydroxypropyl cellulose (2.3:1 ratio) dissolved in 85.5% acetone and 9.5% water (9: 1 ratio). The final dry target of 8 wt% coating will be -20 mg of dried coating per tablet core (on a 250 mg core) to obtain the specified 80% released in 14 hours by USP dissolution in 270 mOsm SIF at pH 6.8 (simulated intestinal fluid). The coated tablets are tray dried for 16 hours at 40°C/30%RH to remove residual solvent. A single 900 um (0.9 mm) delivery orifice is then drilled through one side of the semi-permeable membrane in the center of the tablet face (does not matter which face of the tablet) which was completed by hand drilling.
Table 7. Latrepirdine 15mqA tablets coated with a 8 wt% duration 1 (14h) osmotic coating

* Volatile, not present in final dosage form
The membrane coating was prepared as a 5% (w/w) solution. The coating solution was applied to the tablet cores using a Vector LDCS-5 pan coater using the following parameters:
Table 8. Coating parameters used for AMT coating in the Vector LDCS-5 pan
coater
Coating Parameter Target Value
Coater type Vector LDCS-5 pan coater
1 .3L fully perforated with 4
Coating pan
baffles
Weight tablet cores (g) 1000
Pan RPM 15
Spray Rate (g/min/gun) 20
Number of spray guns 1
Gun-to-Bed distance (inches) 2.5
Atomization Air (PSI) 20
Pattern Air (PSI) 10
Inlet Temperature (°C) 37
Outlet Temperature (°C) 27
Pan Air Flow (CFM) 40
Total solution sprayed for 8
1800
wt% coat (g)
EXAMPLE 18
Small Scale (~1 kg) Osmotic CR Semi-permeable membrane with a hole- Duration 2 (T«n=24h) Tablet Coating Process on Latrepirdine 15mgA Tablet
Cores
Latrepirdine di-hydrochloride, 250mg (15mgA) tablet cores were coated by a functional solvent based film coating spraying process using a Vector LDCS-5 pan coater at small scale (1 kg) with an osmotic polymeric film coating with cellulose acetate as the polymer and hydroxypropyl cellulose as the plasticizer. The acetone and water are the co-solvents uses to dissolve the solids and facilitate pore formation. The coating solution composition contains 5% solids by weight, comprised of 3.75% cellulose acetate and 1.25% hydroxypropyl cellulose (3:1 ratio) dissolved in 85.5% acetone and 9.5% water. The final dry target of 12 wt% coating
will be -30 mg of dried coating per tablet core (on a 250 mg core) to obtain the specified 80% released in 24 hours by USP dissolution in 270 mOsm SIF at pH 6.8 (simulated intestinal fluid). The coated tablets are tray dried for 16 hours at 40°C/30%RH to remove residual solvent. A single 900 um (0.9 mm) delivery orifice is then drilled through one side of the semi-permeable membrane in the center of the tablet face (does not matter which face of the tablet) which was completed by hand drilling.
Table 9. Latrepirdine 15mqA tablets coated with a 12 wt% duration 2 (24h) osmotic coating
Composition of duration 2 coated tablet % in mg tablet coat w/w
coating %)
Latrepirdine Tablet Core 250.0
Cellulose Acetate (Type 398-10) 3.75 22.5 9.0
Hydroxypropyl Cellulose (Klucel EF) 1.25 7.5 3.0
Acetone 85.5 (513)*
Purified water 9.5 (57)*
Total Weight 100% 280.0 12.0
* Volatile, not present in final dosage form
The membrane coating was prepared as a 5% (w/w) solution. The coating solution was applied to the tablet cores using a Vector LDCS-5 pan coater using the following parameters:
Table 10. Coating parameters used for AMT coating in the Vector LDCS-5 pan
coater
Coating Parameter Target Value
Coater type Vector LDCS-5 pan coater
Coating pan 1.3L fully perforated with 4 baffles Weight tablet cores (g) 1000
Pan RPM 15
Spray Rate (g/min/gun) 20
Number of spray guns 1
Gun-to-Bed distance (inches) 2.5
Atomization Air (PSI) 20
Pattern Air (PSI) 10
Inlet Temperature (°C) 37
Outlet Temperature (°C) 27
Pan Air Flow (CFM) 40
Total solution sprayed for 12wt%
2700
coat(g)
Example 19
Hydrophilic matrix controlled release tablet
The metal surfaces of a 25L bin are pre-coated by adding the batch quantity (also see Table 11 below), 2968g, of Avicel (PH 102) and blending for 1 minute at 12 +/- 1 RPM. One-half the batch quantity of Methocel, 1800g, and the Cabosil, 40g, is added to a 25L sized plastic charge bag and gently mixed together by inverting the bag. The 1312g of latrepirdine 2HCI, is added to the center of the bin, meanwhile saving the container it is weighed in. The latrepirdine 2HCI container is rinsed twice with portions of the remaining Methocel, which is subsequently added to the bin. The contents of the plastic charge bag are emptied into the bin, covering the latrepirdine 2HCI. The plastic charge bag is saved. All of the components are blended in the bin for 10 minutes at 12 +/- 1 RPM.
The blend is passed through a Comil rotary mill equipped with a 0.55" screen and a round edge impeller running at approximately 750 RPM. The blend is collected in the saved plastic charge bag. The bin and mill are flushed to remove any residual API by adding the remaining mannitol to the bin, blending for 1 minute at 12 +/- 1 RPM, then releasing contents through the mill into the plastic charge bag. The milled blend is then transferred back to the bin blender. The plastic charge bag is saved. The bin contents are blended for 10 minutes at 12 +/- 1 RPM.
Intragranular magnesium stearate, 40g**, is added to the bin and blended for 5 minutes at 12 +/- 1 RPM. The lubricated blend is processed through a Gerteis roller compactor equipped with knurled rollers, side rims, and an inline oscillating mill containing a pocket rotor and a 0.8mm rasping plate. The target ribbon solid fraction is 0.71 (0.70-0.75) and granules are collected in the saved plastic charge bag.
The collected granules are transferred back to the bin blender and blended for 10 minutes at 12 +/- 1 RPM. Extragranular magnesium stearate, 40g**, is added to the bin and contents are blended for 5 minutes at 12 +/- 1 RPM. Final blend is
affixed above a Kilian T-100 rotary tablet press. Tablets are compressed using 13/32" SRC tooling, to an average target weight of 400 mg +/- 5% and a target hardness of 7 kP. Tablets are passed through a deduster and a metal detector.
** A yield calculation is performed. Intragranular and extra granular magnesium stearate level should be adjusted if yield is less than 98%.
Table 11. 60 mg Latrepirdine hvdrophilic matrix tablet composition; Total tablet weight 400 mg
Ingredient Function % Composition Grams
Latrepirdine* Active ingredient 16.4% 1312
Methocel K4M CR ^. ^™
former providing 45% 3600 Premium Grade
controlled release
Microcrystalline
cellulose, Filler 37.1 % 2968 Avicel PH 102
Colloidal Silicon
Dioxide, Improve Flow 0.5% 40 Cab-O-Sil
Magnesium stearate,
Lubricant 0.5% 40 vegetable grade (IG)
Magnesium stearate,
Lubricant 0.5% 40 vegetable grade (EG)
Total 100% 8000
* Assumes 91 .7% potency factor
(calculating as the free base plus dihyrochloride minus dihydrate)
Example 20
Osmotic Bi-layer swellable core technology (SCT) controlled release tablet Drug Layer
Weight out the API, Polyox N80, and intragranular magnesium sterate according to the batch size (see composition table 12). Add the API and Polyox N80 to an appropriate sized bin blender. Blend for 15 minutes. Add the intragranular magnesium sterate to the blender. Blend for 5 minutes. The lubricated blend is processed through a roller compactor equipped with knurled rollers, side rims, and an
inline oscillating mill containing a pocket rotor and a 0.8mm rasping plate. The target ribbon solid fraction is 0.7-0.75.
The collected granules are weighed to calculate the correct amount of extragrannular magnesium sterate which is then weighed out. The granules are transferred back to the bin blender and blended for 10 minutes at 12 +/- 1 RPM. The extragranular magnesium stearate is added to the bin and contents are blended for 5 minutes at 12 +/- 1 RPM.
Table 12. Latrepirdine SCT drug layer composition; Drug layer target weight for 60 mg dose, 330 mg
Ingredient Function % Composition Grams
Latrepirdine* Active ingredient 20% 200
Polyox N80 Entrainer 79.5% 795
Magnesium stearate, . . . , 1-0/ r
^ Lubricant 0.25% 2.5
vegetable grade (IG)
Magnesium stearate, . . . , 1-0/ r
^ Lubricant 0.25% 2.5
vegetable grade (EG)
Total 100% 1000
* Assumes 91 .7% potency factor
(calculating as the free base plus dihyrochloride minus dihydrate)
Sweller Layer
Weight out the Polyox coagulant, sodium chloride, blue lake dye, and magnesium state according to batch size (see composition Table 13). Add Polyox Coagulant and sodium chloride to appropriate sized bin blender. Sieve blue lake dye through a 20 mesh screen for color uniformity and add to the blender. Blend for 15 minutes.
Table 13. Latrepirdine SCT swell layer composition; swell layer target weight for 60 mg active dose or 330 mg active layer total weight is 165 mg.
Ingredient Function % Composition Grams
Polyox Coagulant Swelling Agent 64.5% 645
Sodium Chloride Osmogen 34.8% 348
Blue Lake #2 Colorant 0.2% 2
Magnesium stearate,
Lubricant 0.5% 5 vegetable grade
Total 100% 1000
Compression
The drug layer and sweller layer are co-compressed using a bi-layer rotary press. Place the drug layer and sweller layer into the two hoppers of a suitable rotary bilayer tablet press, using the drug layer as the first filled layer. The press should be set up with 13/32" standard round concave (SRC) tooling. Compress the drug layer only until the desired fill weight (330 mg for a 60 mg dose) is achieved. Compress the drug layer and sweller layer to target weight of 495 mg. The compression of the active layer to swell layer is completed such that the ratio of the two layers is 2: 1 respectively. The target first layer hardness value is < 1 kp, target overall tablet hardness is 6-8 kp.
Coating and Laser Drilling
Latrepirdine di-hydrochloride, 495 mg (60mgA) tablet cores are coated by a functional solvent based film coating spraying process using a Vector LDCS-5 pan coater at small scale (1 kg) with an osmotic polymeric film coating with cellulose acetate as the polymer and polyethylene glycol 3350 as the plasticizer. The acetone and water are the co-solvents uses to dissolve the solids and facilitate pore formation. The coating solution composition contains 5% solids by weight, comprised of 4.5% cellulose acetate and 0.5% polyethylene glycol 3350 (9: 1 ratio) dissolved in 85.5% acetone and 9.5% water (9: 1 ratio; see Table 14). The final dry target of 22 wt% coating will be -108.9 mg of dried coating per tablet core (on a 495 mg core) to obtain the specified 80% released in 14 hours by USP dissolution in 270 mOsm SIF at pH 6.8 (simulated intestinal fluid). See Table 15 for the coating conditions. The coated tablets are tray dried for 16 hours at 40°C/30%RH to remove residual solvent. A single 900 um (0.9 mm) delivery orifice is then drilled through the drug side of the bilayer tablet (the side which does not contain colorant). The delivery orifice can be drilled by hand or by laser drilling. Table 14. Latrepirdine 60 mgA tablets coated with a 22 wt% (~ 14h T80) osmotic coating

* Volatile, not present in final dosage form
The membrane coating is prepared as a 5% (w/w) solution. The coating solution is applied to the tablet cores using a Vector LDCS-5 pan coater using the following parameters:
Table 15. Coating parameters for SCT coating in the Vector LDCS-5 pan coater
Coating Parameter Target Value
Coater type Vector LDCS-5 pan coater
Coating pan 1.3L fully perforated with 4 baffles
Weight tablet cores (g) 1000
Pan RPM 15
Spray Rate (g/min/gun) 20
Number of spray guns 1
Gun-to-Bed distance (inches) 2.5
Atomization Air (PSI) 20
Pattern Air (PSI) 10
Inlet Temperature (°C) 37
Outlet Temperature (°C) 27
Pan Air Flow (CFM) 40
Total solution sprayed for 22 wt% 4340
coat (g)
Example 21
Pulsatile Beads: Drug Layer Bead
A drug layer solution is prepared by adding 1246.67g purified water to a container equipped with a mixer. With vigorous mixing, 20. Og hydroxypropyl methylcellulose (Methocel E5), and 200. OOg latrepirdine hydrochloride are dissolved therein. Mixing is continued until complete dissolution is achieved.
Microcrystalline Cellulose (MCC) seeds (1000g, 20/25 mesh,) are dispensed into a Wurster fluid bed coater (Glatt 1 .1 ). After fluidisation of the MCC seeds, spraying of the drug layer solution is commenced to layer drug solution effectively onto the seeds. Spraying is continued until the drug layer solution is exhausted (approximately 22% w/w weight gain). The beads are then sieved through a 710μιτι screen and an 850μιτι screen to remove any agglomerated or broken beads.
An isolation layer solution is prepared by adding 664.0 g of water to a container equipped with a mixer. With vigorous mixing, 50. Og hydroxypropyl methylcellulose (methocel E5) is mixed until a complete solution is achieved.
The drug layer cores are dispersed into a Wurster fluid bed coater (Glatt 1.1 ) after fluidisation of the drug layered cores, spraying of the isolation layer is
commenced to layer the isolation solution onto the cores. Spraying is continued until all the solution is exhausted (approximately a 5% w/w weight gain). The beads are dried under the same conditions for 5 minutes. The beads are then sieved through a 710μιτι screen and an 850μιτι screen to remove any agglomerated or broken beads.
A controlled release layer is prepared by 1461 .43g of ethanol to a container equipped with a mixer. With vigorous mixing, 10. Og of trethyl citrate and 100g of methacrylic acid co-polymer type A (Eudragit L100-55) is mixed until a complete solution is achieved.
The drug layer cores are dispersed into a Wuster fluid bed coater (Glatt1.1 ) after fluidisation of the drug layer beads, spraying of the controlled release layer is commenced to layer the controlled release solution on to the beads. Spraying is continued until all the solution is exhausted (approximately 1 1 % w/w weight gain). The beads are dried under the same conditions for 10 minutes. The beads are then sieved through a 710μιτι screen and an 850μιτι screen to remove any agglomerated or broken beads.
The beads are to be transferred to trays and annealed in a temperature and relative humidity controlled oven at 40°C/75% RH for at least 24 hours.
Example 22
Pulsatile Beads: Drug layered Bead
A drug layer solution is prepared by adding 1246.67g purified water to a container equipped with a mixer. With vigorous mixing, 20. Og hydroxypropyl methylcellulose (Methocel E5), and 200. OOg latrepirdine hydrochloride are dissolved therein. Mixing is continued until complete dissolution is achieved.
Microcrystalline Cellulose (MCC) seeds (1000g, 20/25 mesh,) are dispensed into a Wurster fluid bed coater (Glatt 1 .1 ). After fluidisation of the MCC seeds, spraying of the drug layer solution is commenced to layer drug solution effectively onto the seeds. Spraying is continued until the drug layer solution is exhausted (approximately 22% w/w weight gain). The beads are then sieved through a 710μιτι screen and an 850μιτι screen to remove any agglomerated or broken beads.
A controlled release layer is prepared by 1461 .43g of ethanol to a container equipped with a mixer. With vigorous mixing, 10. Og of trethyl citrate and 100g of
methacrylic acid co-polymer type A (Eudragit L100-55) is mixed until a complete solution is achieved.
The drug layer cores are dispersed into a Wuster fluid bed coater (Glatt1.1 ) after fluidisation of the drug layer beads, spraying of the controlled release layer is commenced to layer the controlled release solution on to the beads. Spraying is continued until all the solution is exhausted (approximately 1 1 % w/w weight gain). The beads are dried under the same conditions for 10 minutes.
The beads are to be transferred to trays and annealed in a temperature and relative humidity controlled oven at 40°C/75% RH for at least 24 hours.
Example 23
Preparation of Sweller Layer Coated Beads
A sweller layer suspension is prepared by adding 1708.82 g of Ethanol (96%) to a suitable container with a mixer. With vigorous mixing add 50.00g of povidone (Kollidon® 90F). Mix until solution is complete. Add 300. OOg of croscarmellose sodium while mixing. Continue to mix until a homogenous suspension is formed.
The drug layer cores as described in Example 21 or example 2 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1 .1 ) after fluidisation of the drug layered cores, spraying of the sweller layer suspension is commenced to layer the sweller layer suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 35% w/w weight gain). The beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850μιτι screen and a 1000μιτι screen to remove any agglomerated or broken beads.
Example 24
Preparation of Bursting Coat Layered Beads
The rupture coat suspension is prepared by adding 1594.29g of isopropyl alcohol to a suitable container with a mixer. Add 398.57g of water and mix until solution is complete. Add 120.0g of ethylcellulose (EC4CP) with stirring, continue to mix until the ethylcellulose has dissolved. To the solution add with vigorous mixing 30. Og of magnesium stearate. Stir until a homogenous suspension is formed.
The sweller layer beads as described in Example 23 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidisation of the drug layered cores, spraying of the rupture coat layer suspension is commenced to layer the rupture layer suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 15% w/w weight gain). The beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850μιτι screen and a 1300μιτι screen to remove any agglomerated or broken beads.
An overcoat suspension is prepared by adding 191 .27g of acetone to a suitable container with a mixer. While mixing 8.60g of water is added. Once the solution is complete, add 12.0g of cellulose acetate (CA398-10) and mix until all the cellulose acetate has dissolved. Once the solution is complete add 3.0g of talc and mix until fully dispersed.
The rupture layer coated beads are dispersed into Wurster fluid bed coater (Glatt 1 .1 ) after fluidisation of the cores, spraying of the overcoat coat suspension is commenced to layer the suspension onto the cores. Spraying is continued until an approximate weight gain of 1.5%w/w is achieved. The beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850μιτι screen and a 1300μιτι screen to remove any agglomerated or broken beads.
Example 25
Preparation of Bursting Coat Layered Beads
The rupture coat suspension is prepared by adding 2125.71 g of isopropyl alcohol to a suitable container with a mixer. Add 531.43g of water and mix until solution is complete. Add 160.0g of ethylcellulose (EC4CP) with stirring, continue to mix until the ethylcellulose has dissolved. To the solution add with vigorous mixing 40. Og of magnesium stearate. Stir until a homogenous suspension is formed.
The sweller layer beads as described in Example 23 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidisation of the drug layered cores, spraying of the rupture coat layer suspension is commenced to layer the rupture layer suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 20% w/w weight gain). The beads are
dried under the same conditions for 10 minutes. The beads are then sieved through an 850μιτι screen and a 1300μιτι screen to remove any agglomerated or broken beads.
An overcoat suspension is prepared by adding 191 .27g of acetone to a suitable container with a mixer. While mixing 8.60g of water is added. Once the solution is complete, add 12.0g of cellulose acetate (CA398-10) and mix until all the cellulose acetate has dissolved. Once the solution is complete add 3.0g of talc and mix until fully dispersed.
The rupture layer coated beads are dispersed into Wurster fluid bed coater (Glatt 1 .1 ) after fluidisation of the cores, spraying of the overcoat coat suspension is commenced to layer the suspension onto the cores. Spraying is continued until an approximate weight gain of 1.5%w/w is achieved. The beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850μιτι screen and a 1300μιτι screen to remove any agglomerated or broken beads.
Example 26
Preparation of Bursting Coat Layered Beads
The rupture coat suspension is prepared by adding 2657.14g of isopropyl alcohol to a suitable container with a mixer. Add 664.28g of water and mix until solution is complete. Add 200. Og of ethylcellulose (EC4CP) with stirring, continue to mix until the ethylcellulose has dissolved. To the solution add with vigorous mixing 50. Og of magnesium stearate. Stir until a homogenous suspension is formed.
The sweller layer beads as described in Example 23 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidisation of the drug layered cores, spraying of the rupture coat layer suspension is commenced to layer the rupture layer suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 25% w/w weight gain). The beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850μιτι screen and a 1300μιτι screen to remove any agglomerated or broken beads.
An overcoat suspension is prepared by adding 191 .27g of acetone to a suitable container with a mixer. While mixing 8.60g of water is added. Once the
solution is complete, add 12.0g of cellulose acetate (CA398-10) and mix until all the cellulose acetate has dissolved. Once the solution is complete add 3.0g of talc and mix until fully dispersed.
The rupture layer coated beads are dispersed into Wurster fluid bed coater (Glatt 1 .1 ) after fluidisation of the cores, spraying of the overcoat coat suspension is commenced to layer the suspension onto the cores. Spraying is continued until an approximate weight gain of 1.5%w/w is achieved. The beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850μιτι screen and a 1300μιτι screen to remove any agglomerated or broken beads.
Example 27
Preparation of Bursting Coat Layered Beads
The rupture coat suspension is prepared by adding 3188.58g of isopropyl alcohol to a suitable container with a mixer. Add 797.14g of water and mix until solution is complete. Add 240. Og of ethylcellulose (EC4CP) with stirring, continue to mix until the ethylcellulose has dissolved. To the solution add with vigorous mixing 60. Og of magnesium stearate. Stir until a homogenous suspension is formed.
The sweller layer beads as described in Example 23 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidisation of the drug layered cores, spraying of the rupture coat layer suspension is commenced to layer the rupture layer suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 30% w/w weight gain). The beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850μιτι screen and a 1300μιτι screen to remove any agglomerated or broken beads.
An overcoat suspension is prepared by adding 191 .27g of acetone to a suitable container with a mixer. While mixing 8.60g of water is added. Once the solution is complete, add 12.0g of cellulose acetate (CA398-10) and mix until all the cellulose acetate has dissolved. Once the solution is complete add 3.0g of talc and mix until fully dispersed.
The rupture layer coated beads are dispersed into Wurster fluid bed coater (Glatt 1 .1 ) after fluidisation of the cores, spraying of the overcoat coat suspension is
commenced to layer the suspension onto the cores. Spraying is continued until an approximate weight gain of 1.5%w/w is achieved. The beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850μιτι screen and a 1300μιτι screen to remove any agglomerated or broken beads.
Example 28
Preparation of Bursting Coat Layered Beads
The rupture coat suspension is prepared by adding 3720. Og of isopropyl alcohol to a suitable container with a mixer. Add 7930. Og of water and mix until solution is complete. Add 280. Og of ethylcellulose (EC4CP) with stirring, continue to mix until the ethylcellulose has dissolved. To the solution add with vigorous mixing 70. Og of magnesium stearate. Stir until a homogenous suspension is formed
The sweller layer beads as described in Example 23 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidisation of the drug layered cores, spraying of the rupture coat layer suspension is commenced to layer the rupture layer suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 35% w/w weight gain). The beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850μιτι screen and a 1300μιτι screen to remove any agglomerated or broken beads.
An overcoat suspension is prepared by adding 191.27g of acetone to a suitable container with a mixer. While mixing 8.60g of water is added. Once the solution is complete, add 12.0g of cellulose acetate (CA398-10) and mix until all the cellulose acetate has dissolved. Once the solution is complete add 3.0g of talc and mix until fully dispersed.
The rupture layer coated beads are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidisation of the cores, spraying of the overcoat coat suspension is commenced to layer the suspension onto the cores. Spraying is continued until an approximate weight gain of 1.5%w/w is achieved. The beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850μιτι screen and a 1300μιτι screen to remove any agglomerated or broken beads.
Example 29
Preparation of Bursting Coat Layered Beads
The rupture coat suspension is prepared by adding 4251 .42g of isopropyl alcohol to a suitable container with a mixer. Add 1062.86g of water and mix until solution is complete. Add 320. Og of ethylcellulose (EC4CP) with stirring, continue to mix until the ethylcellulose has dissolved. To the solution add with vigorous mixing 80. Og of magnesium stearate. Stir until a homogenous suspension is formed
The sweller layer beads as described in Example 23 (approximately 1 kg) are dispersed into Wurster fluid bed coater (Glatt 1.1 ) after fluidisation of the drug layered cores, spraying of the rupture coat layer suspension is commenced to layer the rupture layer suspension onto the cores. Spraying is continued until all the suspension is exhausted (approximately a 40% w/w weight gain). The beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850μιτι screen and a 1300μιτι screen to remove any agglomerated or broken beads.
An overcoat suspension is prepared by adding 191 .27g of acetone to a suitable container with a mixer. While mixing 8.60g of water is added. Once the solution is complete, add 12.0g of cellulose acetate (CA398-10) and mix until all the cellulose acetate has dissolved. Once the solution is complete add 3.0g of talc and mix until fully dispersed.
The rupture layer coated beads are dispersed into Wurster fluid bed coater (Glatt 1 .1 ) after fluidisation of the cores, spraying of the overcoat coat suspension is commenced to layer the suspension onto the cores. Spraying is continued until an approximate weight gain of 1.5%w/w is achieved. The beads are dried under the same conditions for 10 minutes. The beads are then sieved through an 850μιτι screen and a 1300μιτι screen to remove any agglomerated or broken beads.
Example 30
Human Clinical Study
The relative bioavailability of 4 different oral sustained release formulations of latrepirdine relative to a single dose of 20 mg latrepirdine immediate release control tablets determining the latrepirdine parent to metabolite Amet AUC ratio was
performed and the following endpoints for latrepirdine and Amet were determined: Cmax, Tmax, AUCo-24 , AUCinf, AUCiast■ An additional endpoint was determined for the relative bioavailability (frei) of latrepirdine for each sustained release formulations relative to the IR formulation.
The study was a randomized, open-label, single dose, 5-period, 5-treatment
10-sequence crossover study in 20 healthy male and female subjects who were CYP2D6 extensive metabolizers (EM) (See table below). Subjects received four different sustained release formulations of latrepirdine and the immediate release control tablet formulation with a washout period of 4 days between doses. All formulations were given as a 20 mg single dose with the exception of SR 3 & 4 which were given as 60 mg.

A: Immediate Release Control Tablet, 20 mg;
B: SR1 , 20 mg;
C: SR2, 20 mg;
D: SR3, 60 mg;
E: SR4, 60 mg.
Subjects were fasted overnight for at least 8 hours prior to administration of the study drug. On the morning of Day 1 of each period, all subjects received a single oral dose of study drug with 240 imL of water. Subjects were allowed a standardized lunch 4 hours after dose administration.
Dosage Forms Administered:
Latrepirdine 20 mg Immediate Release Control Tablet (reference): Preparation A in above Examples.
Latrepirdine 20 mg Sustained Release dosage forms (SR1 and SR2): The preparation of these dosage forms is described in Examples 1 , 2 and 4 respectively (above).
Latrepirdine 60 mg, Sustained Release dosage forms (SR3 and SR4): The preparation of these dosage forms is described in Examples 14, 15 and 16 respectively (above). Four latrepirdine 15 mg SR tablets were given to achieve the 60 mg dose.
During all study periods, blood samples to provide plasma for pharmacokinetic analysis was collected at periodic time points. PK samples were analyzed using standard validated analytical methods. Dose normalized natural log transformed AUC0-24h, AUCinf, AUCiast and Cmax were analyzed for latrepirdine and analyte (when applicable) using a mixed effect model with sequence, period and treatment as fixed effects and subject within sequence as a random effect. Estimates of the adjusted mean differences (Test-Reference) and corresponding 90% confidence intervals were obtained from the model. The adjusted mean differences and 90% confidence intervals for the differences was exponentiated to provide estimates of the ratio of adjusted geometric means (Test/Reference) and 90% confidence intervals for the ratios. The immediate release control tablet formulation was the Reference treatment and the sustained release formulations were the Test treatments.
The relative bioavailability of each latrepirdine SR formulation was determined by calculating the individual ratio of the dose normalized mean area under the plasma concentration versus time curve of the latrepirdine sustained release test formulation (i.e. AUC0-inf) to the dose normalized mean area under the plasma concentration versus time curve of the latrepirdine immediate release reference formulation (i.e. AUC0-inf)- The corresponding individual ratios are then averaged together. The relative bioavailability of the four modified release formulations SR1 , SR2, SR3, and SR4, relative to the IR formulation, were approximately 79%, 88%, 118% and 123%, respectively.
The parent to metabolite AUC0-inf, AUCo-24 and AUCiast ratio for Amet were listed and summarized descriptively by treatment as appropriate. The ratios of AUC0-inf, AUC0-24 and AUCiast were analyzed using a mixed effect model with sequence, period and treatment as fixed effects and subject within sequence as a random effect. Estimates of the adjusted mean differences between the SR and IR formulations, and corresponding 90% confidence intervals was obtained from the model.
The PK parameters AUCo-24 , AUCinf, AUCiast, Cmax, Tmax, and tV2 were summarized descriptively by treatment and analyte (when applicable). For AUCinf and Cmax, individual subject parameters were plotted by treatment for each analyte separately (when applicable). Concentrations were listed and summarized descriptively by PK sampling time, treatment and analyte (when applicable). Individual subject, mean and median profiles of the concentration-time data were plotted by treatment and analyte (when applicable). For summary statistics, and mean and median plots by sampling time, the nominal PK sampling time were used , for individual subject plots by time, the actual PK sampling time was used .
Summary of Plasma latrepirdine Pharmacokinetic Parameter Following Single Oral Doses
Parameter Summary Statistics" by Treatment
Parameter, IR Tablet SRI 20 mg SR2 20 mg SR3 60 mg SR4 60
Units 20 mg mg
N, n 19,15 19,16 19,11 19,19 19,16
AUCmf, 22.33 (67) 15.62 (82) 26.56 (46) 59.86 (69) 59.99 ng*hr/mL (65)
AUClast, 15.05 (85) 13.00 (90) 13.84 (80) 57.86 (70) 57.23 ng*hr/mL (66)
AUC24, 14.89 (85) 12.70 (89) 13.43 (78) 48.02 (73) 40.42 ng*hr/mL (69)
2.751 (96) 1.981 (90) 2.153 (77) 5.005 (76) 3.115
Cmax , ng/mL (71)
2.00 (1.00-4.02) 4.00 (2.00-6.00) 5.00 (3.00-7.00) 6.00 (5.00-8.00) 10.0
(7.00-
T hr 24.0) t½, hr 5.771 (32) 6.093 (31) 6.819 (31) 9.067 (22) 10.23
(21) a Geometric mean (%CV) for all except: median (range) for Tmax; arithmetic mean (%CV) for t½. N = Number of subjects in the treatment group; n = number of subjects where t½ and AUCinf were determined.
IR Tablet = immediate release control tablet (reference); SRI = Eudragit L enteric coated
multiparticulates, examples 1 & 2; SR2 = Eudragit FS enteric coated multiparticulate, example 1 & 4; SR3 = short duration osmotic tablet, examples 14 & 15; SR4 = long duration osmotic tablet, examples 14 & 16
Summary of Plasma A Metabolite Pharmacokinetic Parameter Values
Following Single Oral Doses
Parameter Summary Statistics3 by Treatment
Parameter, IR Tablet SRI 20 mg SR2 20 mg SR3 60 mg SR4 60 Units 20 mg mg
N, n° 19,15 19,15 19, 14 19,18 19,17
682.2 (25) 659.3 (22) 668.8 (21) 2054 (19) 2232 (20)
AUCmf,
ng*hr/mL
660.3 (24) 654.5 (23) 600.0 (25) 1965 (20) 21 13 (18)
AUClast,
ng*hr/mL
636.0 (24) 620.1 (23) 560.6 (24) 1652 (21) 1483 (17)
AUC24,
ng*hr/mL
144.3 (30) 109.3 (27) 101.2 (32) 147.4 (25) 105.3 (18)
Cmax , ng/mL
2.00 (1.00- 3.00 (2.00- 4.00 (3.00-7.00) 5.03 (4.00-10.0) 12.0 3.00) 6.00) (10.0-
 4)
a Geometric mean (%CV) for all except: median (range) for Tmax; arithmetic mean (%CV) for t½. N = Number of subjects in the treatment group; n = number of subjects where t½ and AUCinf were determined.
4)
a Geometric mean (%CV) for all except: median (range) for Tmax; arithmetic mean (%CV) for t½. N = Number of subjects in the treatment group; n = number of subjects where t½ and AUCinf were determined.
IR Tablet = immediate release control tablet (reference); SRI = Eudragit L enteric coated multiparticulates, examples 1 & 2; SR2 = Eudragit FS enteric coated multiparticulate, example 1 & 4; SR3 = short duration osmotic tablet, examples 14 & 15; SR4 = long duration osmotic tablet, examples 14 & 16
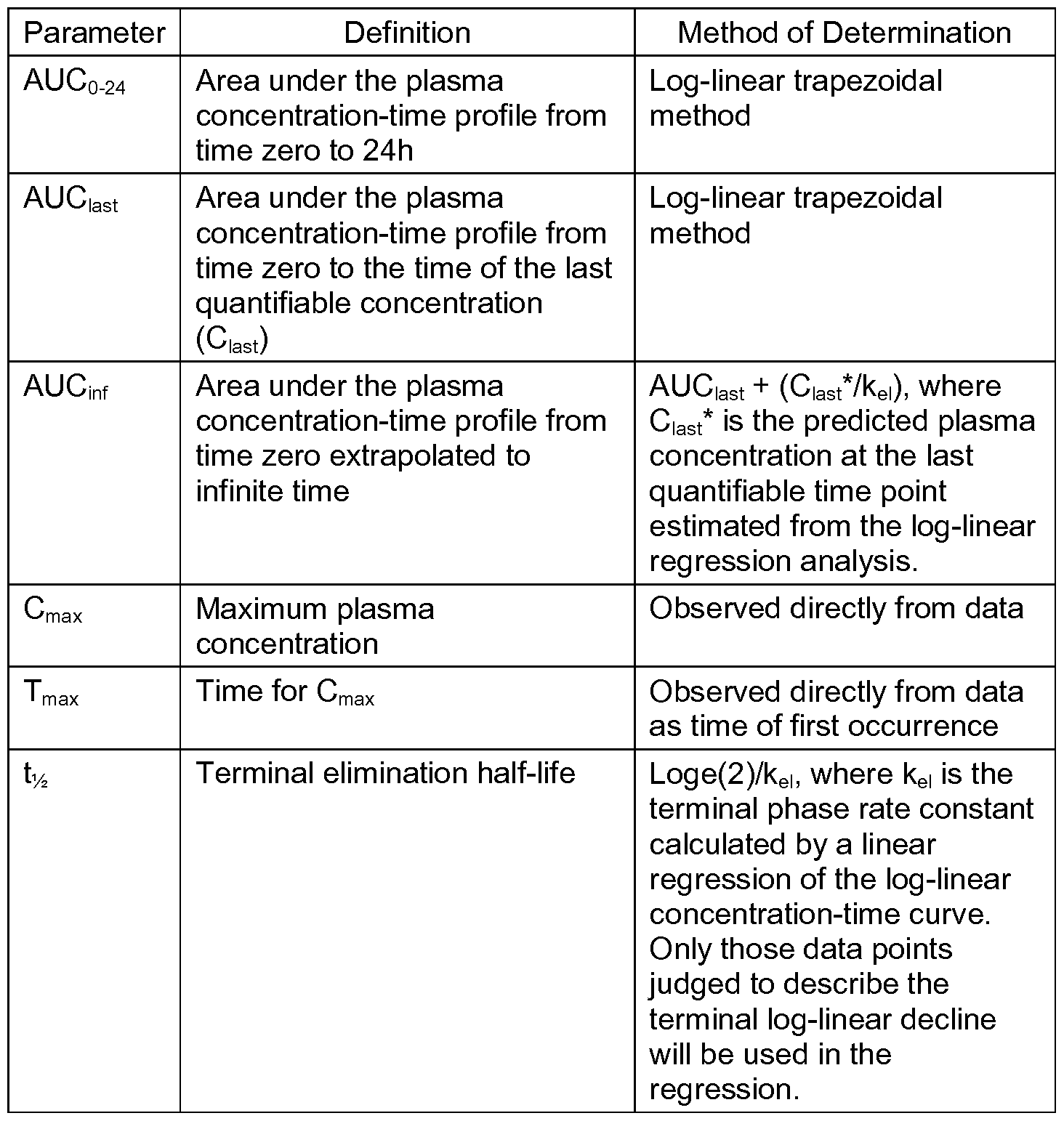
EXAMPLE 31
Latrepirdine Tablet High Drug Loading Osmotic Cores
The metal surfaces of a 20L bin were pre-coated by adding the batch quantity (also see Table 16 below), 554.2 g, of Avicel (PH 200) and blending for 1 minute at 12 +/- 1 RPM. The Cab-o-sil, 12.5g, and one-half the batch quantity of mannitol,
554.2 g, were added to the 20L bin. The 800.0 g of latrepirdine 2HCI, was added to the center of the bin after hand sieving through a #30 mesh sieve (600 micron), meanwhile saving the container it was weighed in. The latrepirdine 2HCI container was rinsed twice with portions of the remaining mannitol, which was subsequently added to the bin. The remaining mannitol was emptied into the bin, covering the latrepirdine 2HCI. All of the components were blended in the bin for 10 minutes at 25 +/- 1 RPM.
The blend was passed through a Comil rotary mill equipped with a 0.042" screen and a round edge impeller running at approximately 300 RPM. The blend was collected in the saved plastic charge bag. The milled blend was then transferred back to the bin blender. The bin contents were blended for 10 minutes at 25 +/- 1 RPM.
Magnesium stearate, 25g, was added to the bin and blended for 3 minutes at 25 +/- 1 RPM. The bin contents were collected in the saved plastic charge bag.
Final blend was affixed above a Kilian T-100 rotary tablet press. Tablets were compressed using 5/16" SRC tooling, to an average target weight of 250. Omg +/- 3% and a target hardness of 7.5 kP. Tablets were passed through a deduster.
Table 16. 80 mqA Latrepirdine Tablet Core Formulation Components
Item # Component Wt % Grams Use
1 Latrepirdine 2HCI ' 32.00 800.00 API
Microcrystalline
2 22.167 554.175 Dliuent cellulose (PH102)
Mannitol 2080,
3 44.333 1108.325 Osmogen gran.
Silicon dioxide
4 0.500 12.500 Glidant (Cab-o-sil)
Magnesium
5 1.000 25.000 Lubricant stearate
Total 100.00 2500.00
* Assumes 92.8% potency
factor
(calculating as the free base plus dihyrochloride
dihydrate)
Claims
1 . A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than about 0.005 but not greater than 0.1 .
2. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose relative bioavailability test/reference crossover study, using a 20 mg latrepirdine immediate release tablet as the reference control, displays a dose normalized relative bioavailability greater than about 1 10%. The immediate release control tablet formulation is the Reference treatment and the sustained release formulations are the Test treatments.
3. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and 12 ng-hr/mL per mg of latrepirdine dosed.
4. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed.
5. A pharmaceutical dosage form comprises latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, as a single dose in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between about 0.78 ng-hr/mL per mg of latrepirdine dosed and about 1.57 ng-hr/mL per mg of latrepirdine dosed and has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.12 ng/ml per mg of latrepirdine dosed.
6. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4.
7. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when added to a test medium comprising 500 mL of 0.2M pH 6.8 potassium phosphate buffer at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 50 rpm, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
8. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than 0.005 but not greater than 0.1 , and when added to a test medium comprising 500 mL of 0.2M pH 6.8 potassium phosphate buffer at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 50 rpm, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
9. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of between 0.36 ng-hr/mL per mg of latrepirdine dosed and12 ng-hr/mL per mg of latrepirdine dosed, and when added to a test medium comprising 500 mL of 0.2M pH 6.8 potassium phosphate buffer at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 50 rpm, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
10. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when added to a test medium comprising 500 imL of 0.2M pH 6.8 potassium phosphate buffer at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 50 rpm, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
1 1. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4, and when added to a test medium comprising 500 imL of 0.2M pH 6.8 potassium phosphate buffer at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 50 rpm, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
12. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when added to a first test medium comprising 500 imL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 imL of a dibasic and monobasic sodium phosphate buffer such that the total phosphate strength is 0.425M, pH 7.6, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
13. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than 0.005 but not greater than 0.1 , and when added to a first test medium comprising 500 imL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 imL of a dibasic and monobasic sodium phosphate buffer such that the total phosphate strength is 0.425M, pH 7.6, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
14. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of at least 0.36 ng- hr/mL per mg of latrepirdine dosed, and when added to a first test medium comprising 500 imL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 imL of a dibasic and monobasic sodium phosphate buffer such that the total phosphate strength is 0.425M, pH 7.6, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
15. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when added to a first test medium comprising 500 imL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 imL of a dibasic and monobasic sodium phosphate buffer such that the total phosphate strength is 0.425M, pH 7.6, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
16. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4, and when added to a first test medium comprising 500 imL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 imL of a dibasic and monobasic sodium phosphate buffer such that the total phosphate strength is 0.425M, pH 7.6, releases 80% of the latrepirdine in no less than 4 hours and no greater than 20 hours.
17. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when added to a first test medium comprising 500 imL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 imL of a dibasic and monobasic sodium phosphate buffer such that the total phosphate strength is 0.425M, pH 7.6, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
18. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than 0.005 but not greater than 0.1 , when added to a first test medium comprising 500 mL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 mL of a dibasic and monobasic sodium phosphate buffer such that the total phosphate strength is 0.425M, pH 7.6, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
19. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of at least 0.36 ng- hr/mL per mg of latrepirdine dosed, when added to a first test medium comprising 500 mL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 mL of a dibasic and monobasic sodium phosphate buffer such that the total phosphate strength is 0.425M, pH 7.6, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
20. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, when added to a first test medium comprising 500 mL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 mL of a dibasic and monobasic sodium phosphate buffer such that the total phosphate strength is 0.425M, pH 7.6, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
21. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4, and when added to a first test medium comprising 500 mL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 mL of a dibasic and monobasic sodium phosphate buffer such that the total phosphate strength is
0.425M, pH 7.6, displays a lag phase of 1 hour in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% or more of the total amount of latrepirdine is released in 2 hours or less.
22. A sustained release pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form, when added to a test medium comprising 500 imL of 0.2M pH 6.8 potassium phosphate buffer at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 50 rpm, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
23. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than 0.005 but not greater than 0.1 , and when added to a test medium comprising 500 mL of 0.2M pH 6.8 potassium phosphate buffer at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 50 rpm, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
24. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of at least 0.36 ng- hr/mL per mg of latrepirdine dosed, and when added to a test medium comprising 500 mL of 0.2M pH 6.8 potassium phosphate buffer at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 50 rpm, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
25. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when added to a test medium comprising 500 imL of 0.2M pH 6.8 potassium phosphate buffer at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 50 rpm, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
26. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4, and when added to a test medium comprising 500 imL of 0.2M pH 6.8 potassium phosphate buffer at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 50 rpm, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
27. A sustained release pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form, when added to a first test medium comprising 500 imL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 imL of 0.425M tribasic sodium phosphate buffer, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
28. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve (AUC0-inf) ratio of latrepirdine to its Amet metabolite which is greater than 0.005 but not greater than 0.1 , and when added to a first test medium comprising 500 imL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 imL of a dibasic and monobasic sodium phosphate buffer such that the total phosphate strength is 0.425M, pH 7.6, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
29. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean area under the plasma concentration versus time curve for the period following administration (AUC0-inf) of at least 0.36 ng- hr/mL per mg of latrepirdine dosed, and when added to a first test medium comprising 500 imL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 imL of a dibasic and monobasic sodium phosphate buffer such that the total phosphate strength is 0.425M, pH 7.6, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
30. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in the fasted state, has a mean maximum plasma concentration (Cmax) of latrepirdine of less than about 0.75 ng/ml per mg of latrepirdine dosed, and when added to a first test medium comprising 500 mL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 mL of a dibasic and monobasic sodium phosphate buffer such that the total phosphate strength is 0.425M, pH 7.6, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
31. A pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier, wherein said dosage form is a sustained release dosage form, and when administered orally to a cohort of subjects, having a CYP2D6 EM status, in a single dose fed / fasted crossover study, has a fed / fasted ratio of the mean area under the plasma concentration versus time curve (AUC0-inf) of 0.7 to 1.4, and when added to a first test medium comprising 500 mL of 0.01 N HCI acid media at 37° C in a standard USP rotating paddle apparatus and the paddles are rotated at 100 rpm followed one hour later by the addition of 400 mL of a dibasic and monobasic sodium phosphate buffer such that the total phosphate strength is 0.425M, pH 7.6, displays a lag phase between 0.5 and 6 hours in which less than 30% of the latrepirdine dose is released, followed by release of latrepirdine at a rate such that 80% of the total amount of latrepirdine is released in no less than 2 hours and no greater than 20 hours.
32. A sustained release pharmaceutical dosage form of Claims 1-31 comprising a core containing latrepirdine and a semi-permeable membrane coating wherein said coating comprises a substantially water-insoluble polymer, and a solid, water-soluble polymeric material.
33. The sustained release dosage form of claim 32 wherein said dosage form delivers latrepirdine primarily by osmotic pressure.
34. The sustained release dosage form of claim 32 wherein said semi- permeable membrane is fabricated by an asymmetric membrane technology.
35. The sustained release dosage form of claim 35 wherein said water insoluble polymer comprises a cellulose derivative.
36. The sustained dosage form of claim 35 wherein said cellulose derivative is cellulose acetate.
37. The sustained release dosage form of claim 32 wherein said solid water soluble polymeric material comprises a polymer having an average molecular weight between 2000 and 50,000 daltons.
38. The sustained release dosage form of claim 37 wherein said solid, water soluble polymeric material is selected from the group consisting of water soluble cellulose derivatives, acacia, dextrin, guar gum, maltodextrin, sodium alginate, starch, polyacrylates, and polyvinyl alcohols.
39. The sustained release dosage form of claim 38 wherein said solid water soluble cellulose derivatives comprises hydroxypropylcellulose, hydroxypropylmethylcellulose or hydroxyethylcellulose.
40. The sustained release dosage form of claim 32 wherein the core also contains a sugar.
41. The sustained release dosage form of claim 40 wherein said sugar is mannitol.
42. A sustained release pharmaceutical dosage form of claims 1-31 comprising latrepirdine and a pharmaceutically acceptable carrier wherein said latrepirdine is embedded in a matrix which releases latrepirdine by diffusion.
43. The sustained release pharmaceutical dosage form of claim 42 wherein a portion of the outside surface of said matrix is covered with an impermeable coating and the remainder of said outside surface is uncovered.
44. The sustained release pharmaceutical dosage form of claim 42 wherein the dosage form is in the form of a tablet and the uncovered surface is in the form of an opening through said impermeable coating.
45. The sustained release pharmaceutical dosage form of claim 42 wherein the dosage form is in the form of a tablet and the uncovered surface is in the form of a passageway which penetrates through the entire tablet.
46. The sustained release pharmaceutical dosage form of claim 42 wherein the dosage form is in the form of a tablet and the uncovered surface is in the form of one or more slits through said impermeable coating or in the form of one or more strips removed therefrom.
47. The sustained release pharmaceutical dosage form of claim 42 wherein the matrix of the dosage form remains substantially intact during the period of latrepirdine release.
48. The sustained release pharmaceutical dosage form of claim 42 wherein the pharmaceutically acceptable carrier comprising the matrix material is selected from the group consisting of waxes, long chain alcohols, fatty acid esters, glycolized fatty acid esters, phosphoglycerides, polyoxyethylene alkyl ethers, long chain carboxylic acids, sugar alcohols, and mixtures thereof.
49. The sustained release pharmaceutical dosage form of claim 42 wherein the outside surface of said matrix is covered with an enteric coating.
50. The sustained release pharmaceutical dosage form of claim 42 wherein the matrix is formed as a melt-congealed core.
51. A sustained release pharmaceutical dosage form of Claims 1-31 comprising latrepirdine and a pharmaceutically acceptable carrier wherein said latrepirdine is embedded in a matrix which releases latrepirdine by eroding.
52. The sustained release pharmaceutical dosage form of claim 51 wherein the matrix of the dosage form comprises hydroxypropyl methylcellulose.
53. The sustained release pharmaceutical dosage form of claim 51 wherein the matrix of the dosage form comprises poly(ethylene oxide).
54. The sustained release pharmaceutical dosage form of claim 51 wherein the matrix of the dosage form comprises polyacrylic acid.
55. A sustained release pharmaceutical dosage form of Claims 1-31 comprising latrepirdine and a pharmaceutically acceptable carrier wherein a reservoir of latrepirdine is encased in a membrane which limits the release rate of latrepirdine by diffusion.
56. The sustained release pharmaceutical dosage form of claim 55 wherein the dosage form is in the form of a tablet coated with a membrane.
57. A sustained release pharmaceutical dosage form of Claims 1-31 comprising latrepirdine and a pharmaceutically acceptable carrier wherein said dosage form is in the form of a multiparticulate comprising particles, which particles are independently coated with a membrane which limits the release rate of latrepirdine by diffusion.
58. The sustained release pharmaceutical dosage form of claim 57 wherein the membrane coating is an enteric membrane.
59. The sustained release pharmaceutical dosage form of claim 57 wherein the membrane coating is a sustained release membrane.
60. A sustained release pharmaceutical dosage form comprising latrepirdine, a pharmaceutically acceptable carrier, and a CYP2D6 inhibitor.
61. A method of treating neurological and psychiatric disorders comprising administering to a patient in need thereof a sustained release pharmaceutical dosage form comprising latrepirdine and a pharmaceutically acceptable carrier in an amount of latrepirdine effective in treating such disorders.
62. A method of treating neurological and psychiatric disorders of claim 61 wherein the sustained release pharmaceutical dosage form is optionally used in combination with a CYP2D6 inhibitor.
63. A method of treating neurological and psychiatric disorders of claim 61 wherein the sustained release pharmaceutical dosage form is optionally used in combination with another active agent.
64. A method for improving the pharmacokinetics of latrepirdine comprising administering to a human in need of such treatment a therapeutically effective amount of a combination of latrepirdine and a CYP2D6 inhibitor or a pharmaceutically acceptable salt thereof.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US24733509P | 2009-09-30 | 2009-09-30 | |
| US61/247,335 | 2009-09-30 | ||
| US37485910P | 2010-08-18 | 2010-08-18 | |
| US61/374,859 | 2010-08-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011039686A1 true WO2011039686A1 (en) | 2011-04-07 |
Family
ID=43066690
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2010/054309 WO2011039686A1 (en) | 2009-09-30 | 2010-09-24 | Latrepirdine oral sustained release dosage forms |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2011039686A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012016707A3 (en) * | 2010-08-06 | 2012-08-30 | Ratiopharm Gmbh | Oral dosage form for the modified release of dimebolin |
| WO2014147526A1 (en) * | 2013-03-16 | 2014-09-25 | Pfizer Inc. | Tofacitinib oral sustained release dosage forms |
| US11786508B2 (en) | 2016-12-31 | 2023-10-17 | Bioxcel Therapeutics, Inc. | Use of sublingual dexmedetomidine for the treatment of agitation |
| US11806429B2 (en) | 2018-06-27 | 2023-11-07 | Bioxcel Therapeutics, Inc. | Film formulations containing dexmedetomidine and methods of producing them |
| US11890272B2 (en) | 2019-07-19 | 2024-02-06 | Bioxcel Therapeutics, Inc. | Non-sedating dexmedetomidine treatment regimens |
| WO2024050028A1 (en) * | 2022-08-31 | 2024-03-07 | Bioxcel Therapeutics, Inc. | Methods and compositions for treating acute stress disorder |
Citations (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA3350A (en) | 1874-04-27 | Franklin Leonard | Machine formaking railway car-coupling pins | |
| CA39810A (en) | 1892-08-12 | James Finney Mcelroy | Valve for train pipes | |
| CA39830A (en) | 1892-08-13 | Alfred Gartner | Gum moistener | |
| US2322793A (en) | 1939-08-21 | 1943-06-29 | Robert S Drummond | Gear finishing tool |
| US3247066A (en) | 1962-09-12 | 1966-04-19 | Parke Davis & Co | Controlled release dosage form containing water-swellable beadlet |
| US3952741A (en) | 1975-01-09 | 1976-04-27 | Bend Research Inc. | Controlled release delivery system by an osmotic bursting mechanism |
| US4519801A (en) | 1982-07-12 | 1985-05-28 | Alza Corporation | Osmotic device with wall comprising cellulose ether and permeability enhancer |
| EP0357369B1 (en) | 1988-08-30 | 1993-05-12 | Pfizer Inc. | The use of asymetric membranes in delivery devices |
| EP0612520A2 (en) * | 1993-02-25 | 1994-08-31 | Pfizer Inc. | PH-triggered osmotic bursting delivery devices |
| US5612059A (en) | 1988-08-30 | 1997-03-18 | Pfizer Inc. | Use of asymmetric membranes in delivery devices |
| US6187785B1 (en) | 1995-10-23 | 2001-02-13 | Selena Pharmaceuticals, Inc. | Agent for treating neurodegenerative disorders |
| WO2004004684A1 (en) * | 2002-07-03 | 2004-01-15 | Pfizer Products Inc. | Controlled-release pharmaceutical formulations |
| WO2005053653A1 (en) | 2003-12-04 | 2005-06-16 | Pfizer Products Inc. | Spray-congeal process using an extruder for preparing multiparticulate azithromycin compositions containing preferably a poloxamer and a glyceride |
| US20050181062A1 (en) | 2003-12-04 | 2005-08-18 | Pfizer Inc | Multiparticulate crystalline drug compositions having controlled release profiles |
| US20050186285A1 (en) | 2003-12-04 | 2005-08-25 | Pfizer Inc | Multiparticulate compositions with improved stability |
| WO2006067576A1 (en) * | 2004-12-21 | 2006-06-29 | Pfizer Products Inc. | Enteric coated azithromycin multiparticulates |
| WO2007057762A2 (en) * | 2005-11-16 | 2007-05-24 | Pfizer Limited | Osmotic bi-layer tablet |
| US20070117834A1 (en) | 2005-10-04 | 2007-05-24 | David Hung | Methods and compositions for treating Huntington's disease |
| US20070225316A1 (en) | 2006-01-25 | 2007-09-27 | Bachurin Sergei O | Methods and compositions for treating schizophrenia |
| US20070248671A1 (en) | 2006-04-24 | 2007-10-25 | Pfizer Inc | Asymmetric membranes for drug delivery devices |
| US20090004281A1 (en) * | 2007-06-26 | 2009-01-01 | Biovail Laboratories International S.R.L. | Multiparticulate osmotic delivery system |
| WO2009111540A1 (en) | 2008-03-04 | 2009-09-11 | Medivation Neurology, Inc. | Methods for preparing pyridylethyl-substituted carbolines |
| EP2236159A2 (en) * | 2009-03-31 | 2010-10-06 | Sanovel Ilac Sanayi ve Ticaret A.S. | Modified release dimebolin compositions |
| EP2236160A2 (en) * | 2009-03-31 | 2010-10-06 | Sanovel Ilac Sanayi ve Ticaret A.S. | Modified release dimebolin formulations |
| WO2010115342A1 (en) * | 2009-04-10 | 2010-10-14 | 中国人民解放军军事医学科学院毒物药物研究所 | SUSTAINED RELEASE COMPOSITION CONTAINING TETRAHYDROPYRIDO [4, 3-b] INDOLE DERIVATIVE AND PREPARATION METHOD OF DERIVATIVE |
-
2010
- 2010-09-24 WO PCT/IB2010/054309 patent/WO2011039686A1/en active Application Filing
Patent Citations (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA3350A (en) | 1874-04-27 | Franklin Leonard | Machine formaking railway car-coupling pins | |
| CA39810A (en) | 1892-08-12 | James Finney Mcelroy | Valve for train pipes | |
| CA39830A (en) | 1892-08-13 | Alfred Gartner | Gum moistener | |
| US2322793A (en) | 1939-08-21 | 1943-06-29 | Robert S Drummond | Gear finishing tool |
| US3247066A (en) | 1962-09-12 | 1966-04-19 | Parke Davis & Co | Controlled release dosage form containing water-swellable beadlet |
| US3952741B1 (en) | 1975-01-09 | 1983-01-18 | ||
| US3952741A (en) | 1975-01-09 | 1976-04-27 | Bend Research Inc. | Controlled release delivery system by an osmotic bursting mechanism |
| US4519801A (en) | 1982-07-12 | 1985-05-28 | Alza Corporation | Osmotic device with wall comprising cellulose ether and permeability enhancer |
| EP0357369B1 (en) | 1988-08-30 | 1993-05-12 | Pfizer Inc. | The use of asymetric membranes in delivery devices |
| US5612059A (en) | 1988-08-30 | 1997-03-18 | Pfizer Inc. | Use of asymmetric membranes in delivery devices |
| US5698220A (en) | 1988-08-30 | 1997-12-16 | Pfizer Inc. | Asymmetric membranes in delivery devices |
| EP0612520A2 (en) * | 1993-02-25 | 1994-08-31 | Pfizer Inc. | PH-triggered osmotic bursting delivery devices |
| US5358502A (en) | 1993-02-25 | 1994-10-25 | Pfizer Inc | PH-triggered osmotic bursting delivery devices |
| US6187785B1 (en) | 1995-10-23 | 2001-02-13 | Selena Pharmaceuticals, Inc. | Agent for treating neurodegenerative disorders |
| WO2004004684A1 (en) * | 2002-07-03 | 2004-01-15 | Pfizer Products Inc. | Controlled-release pharmaceutical formulations |
| WO2005053653A1 (en) | 2003-12-04 | 2005-06-16 | Pfizer Products Inc. | Spray-congeal process using an extruder for preparing multiparticulate azithromycin compositions containing preferably a poloxamer and a glyceride |
| US20050181062A1 (en) | 2003-12-04 | 2005-08-18 | Pfizer Inc | Multiparticulate crystalline drug compositions having controlled release profiles |
| US20050186285A1 (en) | 2003-12-04 | 2005-08-25 | Pfizer Inc | Multiparticulate compositions with improved stability |
| US20080199527A1 (en) | 2004-12-21 | 2008-08-21 | Pfizer Inc. | Enteric Coated Azithromycin Multiparticulates |
| WO2006067576A1 (en) * | 2004-12-21 | 2006-06-29 | Pfizer Products Inc. | Enteric coated azithromycin multiparticulates |
| US20070117834A1 (en) | 2005-10-04 | 2007-05-24 | David Hung | Methods and compositions for treating Huntington's disease |
| US20070117835A1 (en) | 2005-10-04 | 2007-05-24 | David Hung | Methods and compositions for treating Huntington's disease |
| WO2007057762A2 (en) * | 2005-11-16 | 2007-05-24 | Pfizer Limited | Osmotic bi-layer tablet |
| US20070225316A1 (en) | 2006-01-25 | 2007-09-27 | Bachurin Sergei O | Methods and compositions for treating schizophrenia |
| US20070248671A1 (en) | 2006-04-24 | 2007-10-25 | Pfizer Inc | Asymmetric membranes for drug delivery devices |
| US20090004281A1 (en) * | 2007-06-26 | 2009-01-01 | Biovail Laboratories International S.R.L. | Multiparticulate osmotic delivery system |
| WO2009111540A1 (en) | 2008-03-04 | 2009-09-11 | Medivation Neurology, Inc. | Methods for preparing pyridylethyl-substituted carbolines |
| EP2236159A2 (en) * | 2009-03-31 | 2010-10-06 | Sanovel Ilac Sanayi ve Ticaret A.S. | Modified release dimebolin compositions |
| EP2236160A2 (en) * | 2009-03-31 | 2010-10-06 | Sanovel Ilac Sanayi ve Ticaret A.S. | Modified release dimebolin formulations |
| WO2010115342A1 (en) * | 2009-04-10 | 2010-10-14 | 中国人民解放军军事医学科学院毒物药物研究所 | SUSTAINED RELEASE COMPOSITION CONTAINING TETRAHYDROPYRIDO [4, 3-b] INDOLE DERIVATIVE AND PREPARATION METHOD OF DERIVATIVE |
Non-Patent Citations (13)
| Title |
|---|
| COLE ET AL., INT. J. PHARM., vol. 231, 2002, pages 83 - 95 |
| DEW ET AL., BR. J. CLIN. PHARMAC., vol. 14, 1982, pages 405 - 408 |
| G. M. EISENBERG: "Ind. Eng. Chem.", vol. 15, 1943, article "Colorimetric determination of hydrogen peroxide", pages: 327 - 328 |
| HERBIG ET AL., J. CONTROLLED RELEASE, vol. 35, 1995, pages 127 - 136 |
| JOURNAL OF CONTROLLED RELEASE, vol. 134, 2009, pages 74 - 80 |
| KHOSLA; DAVIS, INT. J. PHARMACEUT., vol. 62, 1990, pages R9 - R11 |
| LEFEBVRE; ATOMIZATION; SPRAYS: "Perry's Chemical Engineers' Handbook", 1989 |
| M. ASHRAF-KHORASSANI ET AL.: "Purification of pharmaceutical excipients with supercritical fluid extraction", PHARM. DEV. TECH., vol. 10, 2005, pages 1 - 10 |
| REMINGTON: "The Science and Practice of Pharmacy", 2000 |
| REMINGTON: "The Science and Practice of Pharmacy", 2006, pages: 950 - 1 |
| WATERMAN ET AL.: "Handbook of Isolation and Characterization of Impurities in Pharmaceuticals", 2003, article "Impurities in Drug Products", pages: 75 - 85 |
| YOSHINO, CAPSUGEL SYMPOSIA SERIES; CURRENT STATUS ON TARGETED DRUG DELIVERY TO THE GASTROINTESTINAL TRACT, 1993, pages 185 - 190 |
| YOSHINO: "Capsugel Symposia Series", CURRENT STATUS ON TARGETED DRUG DELIVERY TO THE GASTROINTESTINAL TRACT, 1993, pages 185 - 190 |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012016707A3 (en) * | 2010-08-06 | 2012-08-30 | Ratiopharm Gmbh | Oral dosage form for the modified release of dimebolin |
| WO2014147526A1 (en) * | 2013-03-16 | 2014-09-25 | Pfizer Inc. | Tofacitinib oral sustained release dosage forms |
| US9937181B2 (en) | 2013-03-16 | 2018-04-10 | Pfizer Inc. | Tofacitinib oral sustained release dosage forms |
| US10639309B2 (en) | 2013-03-16 | 2020-05-05 | Pfizer Inc. | Tofacitinib oral sustained release dosage forms |
| EP2968155B1 (en) | 2013-03-16 | 2021-03-03 | Pfizer Inc | Tofacitinib oral sustained release dosage forms |
| US11253523B2 (en) | 2013-03-16 | 2022-02-22 | Pfizer Inc. | Tofacitinib oral sustained release dosage forms |
| US11786508B2 (en) | 2016-12-31 | 2023-10-17 | Bioxcel Therapeutics, Inc. | Use of sublingual dexmedetomidine for the treatment of agitation |
| US11839604B2 (en) | 2016-12-31 | 2023-12-12 | Bioxcel Therapeutics, Inc. | Use of sublingual dexmedetomidine for the treatment of agitation |
| US11931340B2 (en) | 2016-12-31 | 2024-03-19 | Bioxcel Therapeutics, Inc. | Use of sublingual dexmedetomidine for the treatment of agitation |
| US11806429B2 (en) | 2018-06-27 | 2023-11-07 | Bioxcel Therapeutics, Inc. | Film formulations containing dexmedetomidine and methods of producing them |
| US11890272B2 (en) | 2019-07-19 | 2024-02-06 | Bioxcel Therapeutics, Inc. | Non-sedating dexmedetomidine treatment regimens |
| WO2024050028A1 (en) * | 2022-08-31 | 2024-03-07 | Bioxcel Therapeutics, Inc. | Methods and compositions for treating acute stress disorder |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11253523B2 (en) | Tofacitinib oral sustained release dosage forms | |
| US6537573B2 (en) | Combination dosage form comprising cetirizine and pseudoephedrine | |
| KR100958045B1 (en) | Pharmaceutical containing 3-3-dimethylamino-1-ethyl-2-methyl-propylphenol and providing delayed release of the active ingredient | |
| US20140242063A1 (en) | Pharmaceutical compositions | |
| JP2013501810A (en) | Pharmaceutical composition | |
| WO2011039686A1 (en) | Latrepirdine oral sustained release dosage forms | |
| WO2005102272A2 (en) | Sustained-release dosage forms for cabergoline | |
| WO2021014360A1 (en) | Oral modified release dosage forms | |
| RU2790166C2 (en) | Oral dosage forms of tofacitinib with continuous release |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10765523 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10765523 Country of ref document: EP Kind code of ref document: A1 |