DE69629387T2 - Dimere indan verbindungen und ihre pharmazeutische verwendung - Google Patents
Dimere indan verbindungen und ihre pharmazeutische verwendung Download PDFInfo
- Publication number
- DE69629387T2 DE69629387T2 DE69629387T DE69629387T DE69629387T2 DE 69629387 T2 DE69629387 T2 DE 69629387T2 DE 69629387 T DE69629387 T DE 69629387T DE 69629387 T DE69629387 T DE 69629387T DE 69629387 T2 DE69629387 T2 DE 69629387T2
- Authority
- DE
- Germany
- Prior art keywords
- indenyl
- enyl
- ind
- groups
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 0 Cc1c(*)c(*)c(C(*)(*)C(*)(C2(*)*)C(c3c4c(*)c(*)c(*)c3*)=C(*)C4(*)O*)c2c1* Chemical compound Cc1c(*)c(*)c(C(*)(*)C(*)(C2(*)*)C(c3c4c(*)c(*)c(*)c3*)=C(*)C4(*)O*)c2c1* 0.000 description 5
- AGEZXYOZHKGVCM-UHFFFAOYSA-N BrCc1ccccc1 Chemical compound BrCc1ccccc1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- PDZPIKFEKFPDBD-UHFFFAOYSA-N CC(CC1=CC2C=CC=C1)(C1=CCc3ccccc13)C2=O Chemical compound CC(CC1=CC2C=CC=C1)(C1=CCc3ccccc13)C2=O PDZPIKFEKFPDBD-UHFFFAOYSA-N 0.000 description 1
- VACCXFSNVXURQH-UHFFFAOYSA-N CC(Cc1c2cccc1)(C(CC1)c3c1cccc3)C2O Chemical compound CC(Cc1c2cccc1)(C(CC1)c3c1cccc3)C2O VACCXFSNVXURQH-UHFFFAOYSA-N 0.000 description 1
- BENIMPRGASRLHD-UHFFFAOYSA-N CCCC(C1)(C(C)c2c1cccc2)C1=CCc2ccccc12 Chemical compound CCCC(C1)(C(C)c2c1cccc2)C1=CCc2ccccc12 BENIMPRGASRLHD-UHFFFAOYSA-N 0.000 description 1
- LIEXEYQMTNAJAH-UHFFFAOYSA-N CCCc1ccc(CC(Cc2ccccc2)(C2O)C3=Cc4ccccc4C3)c2c1 Chemical compound CCCc1ccc(CC(Cc2ccccc2)(C2O)C3=Cc4ccccc4C3)c2c1 LIEXEYQMTNAJAH-UHFFFAOYSA-N 0.000 description 1
- OJCMELGHKKCEAR-UHFFFAOYSA-N Cc(cc1CC2C3(Cc4ccccc4C3)OC)c(C)c(C)c1C2=O Chemical compound Cc(cc1CC2C3(Cc4ccccc4C3)OC)c(C)c(C)c1C2=O OJCMELGHKKCEAR-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/15—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings
- C07C311/20—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to a carbon atom of a ring other than a six-membered aromatic ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/08—Bronchodilators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C13/00—Cyclic hydrocarbons containing rings other than, or in addition to, six-membered aromatic rings
- C07C13/28—Polycyclic hydrocarbons or acyclic hydrocarbon derivatives thereof
- C07C13/32—Polycyclic hydrocarbons or acyclic hydrocarbon derivatives thereof with condensed rings
- C07C13/45—Polycyclic hydrocarbons or acyclic hydrocarbon derivatives thereof with condensed rings with a bicyclo ring system containing nine carbon atoms
- C07C13/465—Indenes; Completely or partially hydrogenated indenes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C215/00—Compounds containing amino and hydroxy groups bound to the same carbon skeleton
- C07C215/42—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having amino groups or hydroxy groups bound to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C215/44—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having amino groups or hydroxy groups bound to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton bound to carbon atoms of the same ring or condensed ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C225/00—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones
- C07C225/20—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones having amino groups bound to carbon atoms of rings other than six-membered aromatic rings of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/30—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by doubly-bound oxygen atoms
- C07C233/32—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by doubly-bound oxygen atoms with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by a carbon atom of a ring other than a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C251/00—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C251/32—Oximes
- C07C251/34—Oximes with oxygen atoms of oxyimino groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals
- C07C251/44—Oximes with oxygen atoms of oxyimino groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals with the carbon atom of at least one of the oxyimino groups being part of a ring other than a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/63—Esters of sulfonic acids
- C07C309/64—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to acyclic carbon atoms
- C07C309/65—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to acyclic carbon atoms of a saturated carbon skeleton
- C07C309/66—Methanesulfonates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C35/00—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring
- C07C35/22—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring polycyclic, at least one hydroxy group bound to a condensed ring system
- C07C35/23—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring polycyclic, at least one hydroxy group bound to a condensed ring system with hydroxy on a condensed ring system having two rings
- C07C35/32—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring polycyclic, at least one hydroxy group bound to a condensed ring system with hydroxy on a condensed ring system having two rings the condensed ring system being a (4.3.0) system, e.g. indenols
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C35/00—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring
- C07C35/48—Halogenated derivatives
- C07C35/52—Alcohols with a condensed ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C43/00—Ethers; Compounds having groups, groups or groups
- C07C43/02—Ethers
- C07C43/18—Ethers having an ether-oxygen atom bound to a carbon atom of a ring other than a six-membered aromatic ring
- C07C43/188—Unsaturated ethers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/45—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by condensation
- C07C45/46—Friedel-Crafts reactions
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/51—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition
- C07C45/511—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition involving transformation of singly bound oxygen functional groups to >C = O groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/62—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by hydrogenation of carbon-to-carbon double or triple bonds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/65—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by splitting-off hydrogen atoms or functional groups; by hydrogenolysis of functional groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/673—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by change of size of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/68—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/68—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
- C07C45/70—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by reaction with functional groups containing oxygen only in singly bound form
- C07C45/71—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by reaction with functional groups containing oxygen only in singly bound form being hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/68—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
- C07C45/72—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by reaction of compounds containing >C = O groups with the same or other compounds containing >C = O groups
- C07C45/74—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by reaction of compounds containing >C = O groups with the same or other compounds containing >C = O groups combined with dehydration
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/657—Unsaturated compounds containing a keto groups being part of a ring containing six-membered aromatic rings
- C07C49/665—Unsaturated compounds containing a keto groups being part of a ring containing six-membered aromatic rings a keto group being part of a condensed ring system
- C07C49/67—Unsaturated compounds containing a keto groups being part of a ring containing six-membered aromatic rings a keto group being part of a condensed ring system having two rings, e.g. tetralones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/657—Unsaturated compounds containing a keto groups being part of a ring containing six-membered aromatic rings
- C07C49/683—Unsaturated compounds containing a keto groups being part of a ring containing six-membered aromatic rings having unsaturation outside the aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/687—Unsaturated compounds containing a keto groups being part of a ring containing halogen
- C07C49/697—Unsaturated compounds containing a keto groups being part of a ring containing halogen containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/753—Unsaturated compounds containing a keto groups being part of a ring containing ether groups, groups, groups, or groups
- C07C49/755—Unsaturated compounds containing a keto groups being part of a ring containing ether groups, groups, groups, or groups a keto group being part of a condensed ring system with two or three rings, at least one ring being a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/40—Unsaturated compounds
- C07C59/76—Unsaturated compounds containing keto groups
- C07C59/86—Unsaturated compounds containing keto groups containing six-membered aromatic rings and other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C65/00—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C65/32—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups containing keto groups
- C07C65/34—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups containing keto groups polycyclic
- C07C65/36—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups containing keto groups polycyclic containing rings other than six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/007—Esters of unsaturated alcohols having the esterified hydroxy group bound to an acyclic carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/013—Esters of alcohols having the esterified hydroxy group bound to a carbon atom of a ring other than a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/66—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety
- C07C69/73—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids
- C07C69/738—Esters of keto-carboxylic acids or aldehydo-carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/76—Esters of carboxylic acids having a carboxyl group bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/76—Esters of carboxylic acids having a carboxyl group bound to a carbon atom of a six-membered aromatic ring
- C07C69/84—Esters of carboxylic acids having a carboxyl group bound to a carbon atom of a six-membered aromatic ring of monocyclic hydroxy carboxylic acids, the hydroxy groups and the carboxyl groups of which are bound to carbon atoms of a six-membered aromatic ring
- C07C69/92—Esters of carboxylic acids having a carboxyl group bound to a carbon atom of a six-membered aromatic ring of monocyclic hydroxy carboxylic acids, the hydroxy groups and the carboxyl groups of which are bound to carbon atoms of a six-membered aromatic ring with etherified hydroxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/02—Systems containing two condensed rings the rings having only two atoms in common
- C07C2602/04—One of the condensed rings being a six-membered aromatic ring
- C07C2602/08—One of the condensed rings being a six-membered aromatic ring the other ring being five-membered, e.g. indane
Description
- Die Erfindung betrifft Indanverbindungen, Zusammensetzungen, die. diese enthalten und ihre pharmakologische Anwendung.
- Erfindungsgemäß wird eine pharmazeutische Zusammensetzung zur Verfügung gestellt, die eine Verbindung von jedweder der folgenden Formeln umfasst:worin

in den Formeln 1 und 3 R3 bis R15 in Formel 2 R3 bis R1
H, Halogen, Hydroxy, Alkoxy, Aryloxy, Acetoxy, Carboxy, Alkylcarbonyl, Hydrocarbonyl, Amino, Amido, Alkylamino, Hydroxylamino, Aralkylgruppen, mono- und polybenzoide Arylgruppen, substituierten Arylgruppen, Alkyl-, enthaltend 1 bis 10 Kohlenstoffatome oder Cycloalkylgruppen, enthaltend drei 3 bis 8 Kohlenstoffe, substituierten Alkyl- oder Cycloalkylgruppen, worin das Alkyl und Cycloalkyl mit einem oder mehreren gleichen oder unterschiedlichen von Halo, Oxo, Hydroxy, Alkoxy, Aryloxy, Acetoxy, Carboxy, Carbonyl, Amino, Amido, Alkylamino, Hydroxylamino, Aralkylgruppen, mono- und polybenzoiden Alkylgruppen substituiert sind, substituierten Alkylgruppen, in den Formeln 1 und 3 R1, 1R1 und in Formel 2 R2, 1R2 jeweils eines oder mehrere von Hydrocarbonyl, Alkylcarbonyl, Wasserstoff, Hydroxy- oder Acetoxy sind,
in Formel 1 jeweils eines oder mehrere von R1, 1R1, R3, 1R3, R10 zusammen Oxo darstellen können;
in Formel 2 jeweils eines oder mehrere von R2, 1R2, R3, 1R3, R14, 1R14 zusammen Oxo darstellen können;
in Formel 3 jeweils eine oder mehrere von
R1, 1R1, R3, 1R3, R14, 1R14 zusammen
Oxo darstellen können;
und die pharmakologisch annehmbaren Salze, Ester, Amide, Solvate und Isomere davon. - Die Erfindung stellt eine wie zuvor definierte Verbindung der Formeln 1 bis 3 zur Verfügung, wobei folgende Verbindungen ausgeschlossen sind:
1-(2-Indenyl)-1-methylindan-2-on;
1-(2-Indenyl)-indan; und
2-(1-Indenyl)-indanon; - Die Erfindung stellt ebenfalls wie oben definierte Verbindungen der Formeln 1 bis 3 per se zur Verfügung.
In Formel 1 stellen R1, 1R1, R3, 1R3 und R10, 1R10 nicht jeweils Oxo dar. In Formel 2 stellen R1, 1R1, R3, 1R3 und R14, 1R14 nicht jeweils Oxo dar. In Formel 3 stellen R1, 1R1, R3, 1R3 und R14, 1R14 nicht zusammen Oxo dar. - Bevorzugt sind in Formel 1 R3 bis R7 und bevorzugt ebenfalls R10 bis R14 Wasserstoff.
- In Formel 1 sind solche Verbindungen wegen ihrer pharmakologischen Aktivität als Mastzellenstabilisatoren besonders bevorzugt, worin:
R1, 1R1 H, OH bedeuten und
R15 Benzyl bedeutet. - Bevorzugt sind in Formel 2 R3 bis R7 und ebenfalls bevorzugt R10 bis R13 Wasserstoff.
- In Formel 2 sind solche Verbindungen wegen ihrer pharmakologischen Aktivität als Mastzellenstabilisatoren besonders bevorzugt, worin:
R2 1R2 H, OH bedeuten und
R15 Benzyl bedeutet. - Bevorzugt bedeuten in Formel 3 R4 bis R7 und bevorzugt ebenfalls R10 bis R13 Wasserstoff.
- In Formel 3 sind solche Verbindungen wegen ihrer pharmakologischen Aktivität als Maststabilisatoren und wegen ihrer entzündungshemmenden Aktivität insbesondere bevorzugt, worin:
R1, 1R1 H, OH bedeuten und
R15 Benzyl bedeutet. - Die Erfindung betrifft die obigen Verbindungen zur Anwendung als Relaxanzien für glatte Muskeln und/oder als Mastzellenstabilisationsmittel und/oder als entzündungshemmende Mittel.
- Die Erfindung betrifft ebenfalls die Verwendung der Verbindungen in Verfahren zur Propylaxe oder Behandlung, um insbesondere eine relaxierende Aktivität der glatten Muskeln und/oder eine Mastzellen stabilisierende Aktivität und/oder eine entzündungshemmende Aktivität zu erreichen.
- Die Erfindung betrifft ebenfalls die Verbindungen per se, die im Anhang 2 aufgeführt sind.
- Allgemeine Reaktionsprozeduren
- 1. Aluminium-tri-tert.-butoxid-Methode für die Synthese von Indan-1-on
- Indan-1-on und Toluol wurden in einen 250 ml Rundkolben gegeben, und die Lösung wurde durch azetrope Destillation getrocknet. In diese Lösung wurde Aluminium-tri-tert.-butoxid gegeben, und man ließ das Reaktionsgemisch für eine Stunde unter Rückfluss. Eine weitere Menge Aluminium-tri.-tert-butoxid wurde hinzugegeben, und man ließ die Reaktion für weitere dreißig Minuten unter Rückfluss.
- Das Reaktionsgemisch wurde abgekühlt, bevor es in Wasser gegossen wurde. Das Produkt wurde unter Verwendung von Ether extrahiert und über Natriumsulfat getrocknet. Bei der Verdampfung des Lösungsmittels wurde das Rohprodukt über die Blitzsäulenchromatographie (Elutionsmittel: Petroleumether : Ether : 9 : 1) gereinigt. Nach der Verdampfung des Elutionsmittels erhielt das Produkt als kristallinen Feststoff.
- Diese Prozedur ist insbesondere für die Synthese von 2-(1'-Indanyliden)-1-indanon unter Verwendung von 1-Indanon als Ausgangsmaterial anwendbar.
- 2. Lithiumdiisopropylamid(LDA)-Alkylierungsreaktion
- Alkylierungen auf der Basis von LDA eines α-β-Enondimers haben sich als ausgezeichneter Weg zu den α-Alkyl-β, α-Enondimeren erwiesen.
- Im Allgemeinen verläuft die experimentelle Prozedur wie folgt. Ein 100 ml Dreihalskolben wurde im Ofen getrocknet und mit einer Scheidewand und einer Stickstoffeinlassleitung versehen. Der Kolben wurde dann evakuiert und mit einem Fön zur Trocknung erhitzt. In diesen Kolben, der mit Stickstoff gefüllt war, wurde das erforderliche Dimer in trockenem THF gegeben. Die Lösung wurde auf –78°C mit einem Bad aus flüssigen Stickstoff/Ethylacetat gekühlt, und es wurde Lithiumdiisopropylamid (LDA) in THF/Heptan/Ethylbenzol gegeben. Nach dem Rühren für 10 Minuten bei –78°C wurde das gewünschte organische Halogenid hinzugegeben, und man ließ die Lösung bis Raumtemperatur für drei Stunden in einer Stickstoffatmosphäre warm werden. In diese Lösung wurden Ether und eine wässrige Ammoniumchloridlösung gegeben. Die organische Schicht wurde isoliert, und die wässrige Schicht wurde mit Ether extrahiert. Die vereinten organischen Extrakte wurden über Natriumsulfat getrocknet, und die Verdampfung des Lösungsmittels ergab ein Öl. Das Rohprodukt wurde über die Blitzsäulenchromatographie gereinigt.
- 3. Herstellen von Dimeren in den Familien 1 bis 3 durch Verknüpfung eines Silylenolethers eines Indonons mit einem Dimethylacetal oder einem cyclischen Acetal des gleichen Indanons oder eines anderen Indanons.
- Diese Verknüpfungsprozedur wurde primär dafür entwickelt, zwei verschiedene Indanone zusammen zu verknüpfen. Allerdings war diese Methodik ebenfalls für die Verknüpfung des gleichen Indanons miteinander erfolgreich. Im Allgemeinen war die experimentelle Prozedur wie folgt.
- In eine gerührte Lösung des Silylenolethers eines bestimmten Indanons zusammen mit dem entsprechenden Dimethylacetal des gleichen Indanons oder eines davon verschiedenen Indanons in Dichlormethan bei –78°C wurde eine katalytische Menge TMS-Triflate gegeben. Man ließ die Lösung bei –78°C für drei Stunden rühren, wonach man sie dann während einer Stunde –50°C erreichen ließ. In diese Lösung wurde dann eine 5%ige Lösung Natriumbicarbonat gegeben. Die organische Schicht wurde isoliert und die wässrige Schicht mit Dichlormethan extrahiert. Die vereinten organischen Schichten wurden mit Natriumsulfat getrocknet. Nach der Verdampfung des Lösungsmittels ließ man das Rohprodukt durch eine Silicasäule gehen, wobei Petroleumether 100%iger Güte zu Petroleumether : Ethylacetat, 100 : 4 eluiert wurde. Nach der Verdampfung des Elutionsmittels erhielt man das Produkt. Die gleiche Prozedur für die Verknüpfung eines Silylenolethers eines Indanonderivats mit einem cyclischen Ketal eines 1-Indanonderivats.
- 4. Entfernung von Methanol zur Bildung des α,β-ungesättigten Ketons
- Diese Prozedur ist primär dafür ausgestaltet worden, um α,β-ungesättigte Ketone aus den entstehenden Methyletherdimeren, die bei der Verknüpfung der Silylenolether und Dimethylacetalen verschiedener Indanone hergestellt werden, zu synthetisieren. Die Reaktionsprozedur war wie folgt.
- Das entsprechende Dimer wurde in Methanol und DCM, 3 : 1 gelöst, und in diese gerührte Lösung wurde Trifluormethansulfonsäure (Triflic acid) gegeben. Man ließ das Reaktionsgemisch für 1 Stunde unter Rückfluss, wonach sich dann ein Niederschlag bildete. Die Lösung wurde dann in einem Eisbad gekühlt, filtriert und der Feststoff, das das entsprechende α,β-ungesättigte Keton war, wurde getrocknet.
- 5. Verknüpfung von 3-Bromindan-1-on an den Silylenolether von Indanonen
- Diese Prozedur wurde insbesondere darauf gerichtet, eine Vielzahl von Indanonen an die Position 3 des Indan-1-ons zu verknüpfen. Keine der anderen Synthesen, die oben zur Verknüpfung der Indanone miteinander beschrieben wurden, schien für diese Transformation geeignet zu sein. Der Erfolg dieser Verknüpfung war primär durch die Wahl der Lewis-Säure (TMS-Triflate wurde verwendet) wegen der Gegenwart der potentiell reaktiven funktionellen Carbonylgruppe am 3-Bromindanon in Gegenwart von Lewis-Säuren bestimmt. Das Reaktionsschema zur Herstellung einer erfindungsgemäßen Verbindung wie folgt:
-

- In eine gerührte Lösung des Silylenolethers eines Indanons und eines 3-Bromindan-1-on-Derivats in Dichlormethan bei –78°C wurde eine katalytische Menge TMS-Triflate gegeben. Man ließ die Lösung bei –78°C für 10 Minuten und bei Raumtemperatur für 3 Stunden rühren. In diese Lösung wurde dann festes Natriumbicarbonat (etwa 2 g) gegeben, und die Lösung wurde schnell für 10 Minuten gerührt. Die Lösung wurde dann filtriert, und das Filtrat wurde eingedampft, wobei ein mobiles Öl zurückgelassen wurde, das durch eine Silicasäule mit Petroleumether : Ethylacetat, 9 : 2 als Lösungsmittel, gelassen wurde. Nach der Verdampfung des Lösungsmittels erhielt man das Produkt.
- 6. Reduktion der Dimere mit 10% Palladium auf Kohlenstoff
- Diese Prozedur ist insbesondere auf die Reduktion der Kohlenstoff-Kohlenstoff-Doppelbindungen von α,β-Enondimeren in den Familien 1, 2 und 3 anwendbar. Im Fall der α,β-ungesättigten Ketonindandimere führt diese Reduktionsmethode immer sowohl zur Reduktion der Kohlenstoff-Kohlenstoff-Doppelbindung und des Carbonyls des α,β-ungesättigten Systems. Die Reduktionsprozedur war wie folgt.
- Das entsprechende Dimer wurde in Ethanol und Ethylacetat gelöst. Dazu wurde 10%iges Palladium über Aktivkohle (katalytische Mengen) zugegeben, und die Reaktion wurde unter Wasserstoff für 2 Stunden gerührt. Der Katalysator wurde durch Filtration entfernt. Die Verdampfung des Lösungsmittels bei reduziertem Druck ergab das Rohprodukt. Das Rohprodukt wurde über die Blitzsäulenchromatographie gereinigt.
- 7. Reduktion der Dimere mit 10% Palladium auf Kohlenstoff und konzentrierter wässriger HCl
- Diese Prozedur ist insbesondere auf die Reduktion der β,α-Kohlenstoff-Kohlenstoff-Doppelbindung und der funktionellen Ketongruppe anwendbar. Die Reduktionsprozedur war wie folgt.
- Das entsprechende Dimer wurde in destilliertem Ethanol und Ethylacetat gelöst. Dazu wurde eine konzentrierte wässrige 37 %ige HCl-Lösung zusammen mit Wasser und 10% Palladium über Aktivkohle (katalytische Mengen) gegeben, und die Mischung wurde unter Wasserstoff für 24 Stunden gerührt.
- Der Katalysator wurde durch Filtration entfernt, und das Produkt wurde in Ethylacetat (3 × 20 ml) extrahiert. Das Rohprodukt wurde über die Blitzsäulenchromatographie gereinigt.
- 8. Natriumboranatreduktion von Dimeren
- Diese Reduktion ist insbesondere auf die Reduktion der funktionellen Ketongruppe der Verbindungen in den Familien 1–4 anwendbar. Die Reduktionsprozedur war wie folgt.
- Das entsprechende Dimer wurde in Ethanol gelöst, und es wurde Natriumboranat in die Reaktion in kleinen Mengen über 10 Minuten gegeben. Die Reaktion wurde dann bei Raumtemperatur für 3 Stunden gerührt. Das Reaktionsgemisch wurde in Wasser (20 ml) gegossen und in Diethylether (3 × 20 ml) extrahiert. Die Blitzsäulenchromatograhie über Silikagel erbrachte das Produkt.
- 9. Reduktion der Dimere durch die Huang-Minlon-Modifikationsreaktion mit der Hydrazinhydratreaktion
- Diese Reduktionsprozedur ist insbesondere auf die Reduktion der funktionellen Ketongruppe im Fall der β,α-Enone anwendbar. Die Reduktionsprozedur war wie folgt.
- Das entsprechende Dimer wurde in Ethylenglykol dispergiert. Es wurde Hydrazinhydrat zusammen mit Natriumhydroxid hinzugegeben. Man ließ die Reaktion für 24 Stunden unter Rückfluss rühren. Das Reaktionsgemisch wurde dann auf Raumtemperatur abgekühlt, und es wurde Wasser hinzugegeben und das Produkt mit Ethylacetat extrahiert. Die organische Schicht wurde isoliert und über wasserfreiem Natriumsulfat getrocknet. Es wurde die Blitzsäulenchromatographie verwendet, um das reine Produkt herzustellen.
- 10. Cyanoborhydridreduktion von Dimeren
- Diese Reduktionsprozedur ist insbesondere auf die Reduktion der funktionellen Ketongruppe der Verbindungen in den Familien 1–3 geeignet. Die Reduktion ist wie folgt.
- Das entsprechende Dimer wurde in 1,2-Dichlorethan bei Raumtemperatur dispergiert. In diese Lösung wurden festes Zinkjodid und Natriumcyanoboranat gegeben. Die Reaktion wurde unter Rückfluss für 20 Stunden gerührt. Das Produkt wurde in Wasser gegeben und in Ethylacetat extrahiert. Es wurde die Blitzsäulenchromatographie (Elutionsmittel: Petroleumether : Ethylacetat, 9 : 1) verwendet, um das reine Produkt zu isolieren.
- 11. Reduktion oder Isomerisierung der α,β-ungesättigten Doppelbindung in Dimeren mit 5% Palladium auf Kohlenstoff
- Reduktion oder Isomerisierung der α,β-ungesättigten Doppelbindung in Dimeren mit 5% Palladium auf Kohlenstoff
- Diese Prozedur ist insbesondere auf die Reduktion der Doppelbindung im Fall der α,β-ungesättigten Ketone anwendbar.
- Das entsprechende Dimer wurde in Ethanol und Ethylacetat dispergiert, und in dieses wurde 5% Palladium auf Kohlenstoff hinzugegeben. Die Mischung wurde unter Wasserstoff für 14 Stunden gerührt. Das Palladium wurde durch Filtration entfernt, und das Lösungsmittel wurde entfernt, um das Rohreaktionsprodukt zu erhalten. Die Blitzsäulenchromatographie ergab das gewünschte Produkt.
- 12. Wilkinson-Reduktion von Dimeren
- Diese Reduktionsmethode war insbesondere für die selektive Reduktion einer Doppelbindung an R15 ohne Reduzierung der R8-R9-Doppelbindung in den Familien 1 bis 3 effektiv. Die Reduktionsprozedur war wie folgt.
- Das jeweilige Dimer wurde in Ethanol und Ethylacetat gelöst. In diese Lösung wurde unter Rühren der Wilkinson-Katalysator hinzugegeben. Die Reaktion wurde dann unter Wasserstoff für 20 Stunden gerührt. Das Produkt war zwischen Dimethylacetat und Wasser aufgeteilt, und die organische Schicht wurde isoliert und mit Na2SO4 getrocknet. Das Rohprodukt wurde über die Blitzsäulenchromatographie gereinigt, und man erhielt das gewünschte Produkt.
- 13. Hydrolyse eines Esterdimers in den Familien 1 bis 3
- Der betreffende Ester wurde in einer Lösung aus 1,45 Mol NaOH in THF : MeOH : H2O (6 : 3 : 2) gelöst, die dann unter Rückfluss gehalten wurde. Nach 20 Minuten zeigte die TLC, dass die Hydroylse des Esters vollständig war. Nach Abkühlen des Reaktionsgemischs wurde eine gesättigte Lösung aus wässrigen Ammoniumchlorid, wässriger HCl (2 Mol) und Ether hinzugegeben. Die orgenische Schicht wurde isoliert, und die wässrige Schicht wurde mit Ether extrahiert. Die vereinten organischen Extrakte wurden mit Na2SO4 getrocknet und filtriert. Nach der Verdampfung des Lösungsmittels blieb die Säure zurück.
- 14. Oximsynthese
- Diese Prozedur ist insbesondere anwendbar auf die Synthese von Oximderivaten der Ketonindandimere, die Wasserstoffe zum Keton aufweisen, anwendbar. Im Allgemeinen war die Prozedur wie folgt.
- Das Ketonindanondimer wurde in einer Lösung aus Methanol : Pyridin (4 : 1) gelöst, und in diese Lösung wurde dann Hydroylaminhydrochlorid gegeben. In Abhängigkeit des spezifischen Ketonindandimers wurde die Reaktion entweder bei Raumtemperatur oder bei Rückflussbedingungen durchgeführt.
- 15. O-Alkylierung des Oxims
- Diese Prozedur ist insbesondere auf die O-Alkylierung des synthetisierten Oximderivate anwendbar. Im Allgemeinen war die Prozedur wie folgt.
- Eine Lösung aus dem Oximindandimer wurde in Ether : tert.-Butanol 3 : 1 gelöst. Es wurde im Allgemeinen Benzylbromid als Alkylierungsmittel verwendet, und es wurde in das Reaktionsgemisch gegeben. Kalium-tert.-butoxid, 1 Äq, wurde tropfenweise in diese Lösung bei Raumtemperatur gegeben. Nach der Aufarbeitung unter Verwendung von wässrigen Ammoniumchlorid und Ether wurde der gewünschte Oximether nach der Chromatographie isoliert.
- 16. α-Alkylierung der O-Benzyloxime
- Diese Prozedur ist insbesondere auf die α-Alkylierung von Oximetherderivaten anwendbar.
- Die Prozedur war wie folgt.
- Eine Lösung aus dem Oximether wurde in trockenem Ether gelöst und auf –78°C heruntergekühlt. Diese Lösung wurde n-Butyllithium gegeben, wonach Benzylbromid im Überschuss folgte. Die Reaktion wurde im Allgemeinen mit Wasser abgeschreckt, das Produkt mit Ether extrahiert und über die Blitzsäulenchromatographie gereinigt.
- 17. Sulfonylierung der 2-Indanoldimere
- Diese Prozedur ist insbesondere auf die Sulfonylierung der Hydroxylgruppen von 2-Indanoldimere anwendbar.
- Das entsprechende hydroxylierte Dimer wurde in Dichlormethan gelöst, und in diese Lösung wurden tropfenweise Methansulfonylchlorid und N,N-Diisopropylethylamin gegeben. Nach dem Rühren für 15 Minuten bei 0°C war das Reaktionsgemisch normalerweise zwischen DM und dem wässrigen NaHCO3 aufgeteilt, die organische Schicht wurde isoliert, mit Wasser, 2 Mol wässriger HCl und schließlich mit Wasser gewaschen. Die Endreinigung der Produkte erfolgte über die Blitzsäulenchromatographie.
- 18. Acetylierung der Hydroxylindandimere
- Im Allgemeinen dient die Prozedur dazu, die Verbindung für die Acetylierung in DCM zu lösen und Essigsäureanhydrid als Acetylierungsreagenz mit Triethylamin als tertiäre Base und DMAP als Acelyierungskatalysator zu verwenden.
- 19. Transformation von β-Methyoxycarbonylverbindungen in die α-Alkyl- und β-Enone
- Die β-Methoxycarbonylverbindungen wurden in Ether : Butanol (5 : 1) gelöst, und in dieses wurde das gewünschte Alkylierungsmittel gegeben. In die Lösung wurde unter Rühren Kalium-tert.-butoxid tropfenweise über einen Zeitraum von 30 Minuten gegeben. Die Reaktion ließ man bei Raumtemperatur für 24 Stunden rühren. Es wurde eine wässrige Lösung aus Ammoniumchlorid hinzugegeben, und das Produkt wurde in Ether extrahiert. Das Rohreaktionsgemisch wurde dann durch eine Blitzsilikasäule gelassen, und man erhielt das gewünschte Produkt.
- 20. Alkylierung eines α,β-Enons
- Das entsprechende Dimer wurde in Ether : tButanol (5 : 1) gelöst, und dazu wurde das gewünschte Alkylierungsmittel gegeben. In die Lösung wurde unter Rühren Kalium-tert.-butoxid tropfenweise über einen Zeitraum von 30 Minuten gegeben. Man ließ die Reaktion bei Raumtemperatur für 24 Stunden rühren. Es wurde eine wässrige Lösung aus Ammoniurnchlorid hinzugegeben, und das Produkt wurde in Ether extrahiert. Das Rohreaktionsgemisch wurde dann durch eine Blitzsilikakolonne gelassen, und man erhielt das gewünschte Produkt.
- Synthese von 1C1 Kalium-tert.-butoxid-Methode

- Es wurden Kalium-tert.-butoxid (4,25 g, 37 mMol) in tButanol (125 ml) und Ether (10 ml) tropfenweise über 20 Minuten in eine Lösung aus Indan-1-on (5,0 g, 37 mMol) in Ether (20 ml) und tButanol (5 ml) unter Rühren gegeben. Man ließ dann das Reaktionsgemisch über Nacht rühren.
- Das Rohprodukt war zwischen dem Ethylacetat und dem gesättigten wässrigen Ammoniumchlorid aufgeteilt. Die organische Schicht wurde isoliert, und die wässrige Phase wurde mit Ethylacetat wieder extrahiert. Die organischen Schichten wurden vereint und über Natriumsulfat getrocknet. Bei der Verdampfung des Lösungsmittels erhielt man das Rohprodukt. Es wurde die Blitzchromatographie angewendet, um das gewünschte Produkt zu reinigen (Elutionsmittel : Petroleumether (Siedepunkt 40–60°C): Ethylacetat, 9 : 1). Bei der Umkristallisierung mit Ether wurde das 1Cl als weißer Feststoff erhalten. (Ausbeute : 20%).
- Massenspektrum geringer Auflösung: gefunden M+246
erforderlich M+246
1H NMR (CDCl3, 300 MHz) δH 3.11 (2H, t, J = 6 Hz, CH 2), 3.54 (2H, m, CH 2), 3.98 (2H, s, CH 2), 7.53 (6H, m, 6 × Ar-H, 7.79 (2H, m, 2 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 30.9, 31.5, 33.0 (3 × CH2), 123.5, 125.9 125.7 (3 × Ar-CH), 125.9 (C=C), 126.2, 126.8, 127.2, 130.4, 133.5 (5 × Ar-CH), 139.5, 140.8, 148.5, 151.7, 154.9 (1 × C=C und 4 × AR-C), 195.1 (C=O). - Alternativsynthese für 1C1 Aluminium-tri.-tert.-butoxid-Methode

- Es wurden Indan-1-on (5,0 g, 37 mMol) und Toluol (80 ml) in einen 250 ml Rundkolben gegeben, und die Lösung wurde über die azeotrope Destillation getrocknet. In diese Lösung wurde Aluminium-tri-tert.-butoxid (4,7 g, 19 mMol) gegeben, und man ließ das Reaktionsgemisch für 1 Stunde unter Rückfluss. Eine weitere Menge Aluminium-tri.-tert.-butoxid (2,3 g, 9,0 mMol) wurde hinzugegeben, und man ließ die Reaktion für weitere 30 Minuten unter Rückfluss.
- Das Reaktionsgemisch wurde gekühlt, bevor es in Wasser gegossen wurde. Das Produkt wurde unter Verwendung von Ether extrahiert und über Natriumsulfat getrocknet. Nach Verdampfung des Lösungsmittels wurde das Rohprodukt über die Blitzsäulenchromatographie (Elutionsmittel: Petroleumether: Ether, 9 : 1) gereinigt. Nach der Verdampfung des Lösungsmittels erhielt man das 1Cl als weißen kristallinen Feststoff, 48% Ausbeute.
Massenspektrum geringer Auflösung: gefunden M+246
Erforderlich M+246
1H NMR (CDCl3, 300 MHz) δH 3.11 (2H, t, J = 6 Hz, CH 2), 3.54 (2H, m, CH 2), 3.98 (2H, s, CH 2), 7.53 (6H, m, 6 × Ar-H), 7.79 (2H, m, 2 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 30.9, 31.5, 33.0 (3 × CH2), 123.5, 125.7, 125.9 (3 × Ar-CH), 125.9 (C=C), 126.2, 126.8, 127.2, 130.4, 133.5 (5 × Ar-CH), 139.5, 140.8, 148.7, 151.7, 154.9 (4 × Ar-C, and 1 × C=C), 195.1 (C=O). - Synthese von 1C2

- Ein 100 ml Dreihalsrundkolben wurde ofengetrocknet und mit einer Scheidewand und einer Stickstoffeinlassleitung versehen. Der Kolben wurde dann evakuiert und mit einem Fön zur Trocknung erhitzt. In diesen Kolben, der mit Stickstoff gefüllt war, wurde das Indan-1-on-Dimer 1Cl (500 mg, 2,0 mMol) in trockenem THF (25 ml) gegeben. Die Lösung wurde auf –78°C mit einem Bad aus flüssigem Stickstoff/Ethylacetat heruntergekühlt, und es wurde Lithiumdiisopropylamid (LDA) in THF/Heptan/Ethylbenzol (1,0 ml einer 2 molaren Lösung aus LDA) hinzugegeben. Nach dem Rühren für 10 Minuten bei –78°C wurde Jodmethan (1,14 g, 8,0 mMol, 4 Äquivalente) hinzugegeben, und man ließ die Lösung auf Raumtemperatur für 3 Stunden unter Vakuum und in einer Stickatmosphäre warm werden.
- In diese Lösung wurden Ether (30 ml) und eine Ammioniumchloridlösung (30 ml) gegeben. Die organische Schicht wurde isoliert, und die wässrige Schicht wurde mit Ether (2 × 30 ml) extrahiert. Die vereinten organischen Extrakte wurden über Natriumsulfat getrocknet, und nach der Verdampfung des Lösungsmittels ergab sich ein Öl. Das Rohprodukt wurde über die Blitzsäulenchromatographie (Elutionsmittel : Petroleumether, Siedepunkt 40 bis 60°C : Ethylacetat, 9 : 1) gereinigt und man erhielt 1C2.
Schmelzpunkt: 112–114°C
IR (KBr)max 2361,2, 1715,1, 1606,9, 1459,7 cm–1
Mikroanalyse : C19H16O erforderlich C, 87,69% und H, 6,15
Gefunden: C, 87,54% und H 6,25
Massenspektrum geringer Auflösung: gefunden: M+260, M+ – 15 = 245
erforderlich: M+260
1H NMR (CDCl3, 300 MHz) δH 1,67(3H, s, CH 3), 3.20 (1H, d, J = 17 Hz, CH), 3.40 (2H, d, J = 2 Hz, CH 2), 3.69 (1H, d, J = 17 Hz, CH), 6.52 (1H, t, J = 2 Hz, CH), 6.87 (1H, d, J = 8 Hz, Ar-H), 7.15 (2H, m, 2 × Ar-H), 7.68 (1H, m, Ar-H), 7.68 (1H, m, Ar-H), 7.92 (1H, d, J = 8 Hz, Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 23.9 (CH3), 37.6, 41.2 (2 × CH2), 50.5 (COC(CH2)(CH3)), 119.8, 124.1, 124.6, 124.8,125.9, 126.8, 127.7, 130.1, 135.2, (8 × Ar-CH & 1 × C=CH), 135.6, 143.0, 144.9, 145.8, 152.3 (4 × Ar-C & C=CH) 208.6 (C=O). - Synthese von 1C2 Kalium-tert.-butoxid-Methode

- Man gab in einen Dreihalsrundkolben Indan-1-on (10,0 g, 75 mMol), das in Ether (100 ml) und tButanol (20 ml) gelöst war. Dazu wurden Jodmethan (4,72 ml, 75 mMol) in Ether (50 ml) und Kalium-tert.-butoxid (8,49 g, 75 mMol) in tButanol (150 ml) tropfenweise bei gleichen Raten gegeben. Die Reaktion wurde unter Rückfluss für 2 Stunden gerührt.
- Man ließ die Lösung abkühlen, und die Mischung war zwischen dem Ethylacetat und dem wässrigen Ammoniumchlorid (1 : 1 300 ml) aufgeteilt. Die organische Schicht wurde extrahiert und die wässrige Phase mit Ethylacetat (2 × 50 ml) wieder extrahiert.
- Die vereinten organischen Schichten wurden über Natriumsulfat getrocknet. Nach der Verdampfung des Lösungsmittels erhielt man das Rohprodukt. Die Blitzsäulenchromatograhie wurde angewendet, um das gewünschte Produkt zu reinigen (Elutionsmittel Petroleumether (Siedepunkt 40–60°C) : Ethylacetat, 9 : 1). Nach der Isolierung von 1C2 wurde dieses aus Ether umkristallisiert, und man erhielt einen weißen Feststoff (20%).
Schmelzpunkt: 112–114°C
IR (KBr)max: 2361,2, 1715,1, 1606,9, 1459,7 cm–1
Mikroanalyse: C19H16O erforderlich C, 87,69% und H, 6,15
gefunden: C, 87,54% und H, 6,25
Massenspektrum geringer Auflösung: gefunden: M+260, M+ – 15 = 245
erforderlich: M+260.
1H NMR (CDCl3, 300 MHz) δH 1.67 (3H, s, CH 3), 3.20 (1H, d, J = 17 Hz, CH), 3.40 (2H, d, J = 2 Hz, CH 2), 3.69 (1H, d, J = 17 Hz, CH), 6.52 (1H, t, J = 2 Hz, CH), 6.87 (1H, d, J = 8 Hz, Ar-H), 7.15 (2H, m, 2 × Ar-H), 7.68 (1H, m, Ar-H), 7.68 (1H, m, Ar-H), 7.92 (1H, d, J = 8 Hz, Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 23.9 (CH3), 37.6, 41.2 (2 × CH2), 50.5 (COC(CH2)(CH3)), 119.8, 124.1, 124.6, 124.8, 125.9, 126.8, 127.7, 130.1, 135.2, (2 × Ar-CH & 1 × C=CH), 135.6, 143.0, 144.9, 145.8, 152.3 (4 × Ar-C & C=CH), 208.6 (C=O). - Reduktion mit 10% Palladium auf Kohlenstoff Synthese von 1C4

- 1C2 (1,0 g, 3,8 mMol) wurde in Ethanol (20 ml) und Ethylacetat (10)ml) gelöst. Dazu wurde 10% Palladium über Aktivkohle (katalytische Mengen) gegeben, und die Reaktion wurde unter Wasserstoff für 2 Stunden gerührt. Der Katalysator wurde durch Filtration entfernt. Die Verdampfung des Lösungsmittels bei reduziertem Druck ergab ein klares Öl, das aus Diethylether und Petroleumether (Siedepunkt 40–60°C) als weißer Feststoff, 0,76 g, 76,34% umkristallisiert wurde. Es wurde festgestellt, dass dieser weiße Feststoff ein Diastereomerengemisch war.
Schmelzpunkt: 88–90°C
IR (Film)max: 1709, 8 cm–1 (C=O) , 1606, 8 cm–1 (C=C). - Bei Unterscheidungen sind die Werte für das kleinere Diastereomere kursiv angezeigt.
1H NMR (CDCl3, 300 MHz) δH 1.46 (3H, s, CH 3), 1.45, 1.90, 2.10 & 2.30 (2H, 4 × m, CHCH 2CH2), 2.68 & 2.98 (2H, dd, J = 17.8 Hz, CCH 2), 2.79 (2H, m, CHCH2CH 2), 3.65, 3.85 (1H, m, CHCH2CH2), 6.75 & 6.96 (1H, br.m & t, 1 × Ar-H), 7.30 (5H, br m, 5 × Ar-H), 7.56 (1H, m, 1 × Ar-H), 7.78, 7.83 (1H, dd, 1 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 24.4, 24.6 (CH3), 29.0 28.3, 31.8, 31.2, 37.6, 36.8 (3 × CH2), 50.7, 50.6 (CH), 52.9, 52.6 (qC), 124.2, 124.8, 125.5, 125.9, 126.4, 126.8, 127.4, 134.8 (Ar-CH), 136.2, 144.2, 145.0, 153.4 (Ar-C), 210.9, 211.0, (C=O). - Reduktion mit 10% Palladium auf Kohlenstoff Synthese von 1C4

- 1C2 (100 mg, 0,385 mMol) wurde in destilliertem Ethanol (5 ml) und Ethylacetat (1 ml) gelöst. In diese Lösung wurde konzentrierte HCl, 37%ige Lösung (0,2 ml,) zusammen mit Wasser (0,4 ml) und Pd/Kohle (katalytische Mengen) gegeben, und die Mischung wurde unter Wasserstoff für 24 Stunden gerührt.
- Der Katalysator wurde durch Filtration entfernt, und das Produkt wurde in Ethylacetat (3 × 20 ml) extrahiert. Das Rohprodukt wurde über die Blitzsäulenchromatographie (Elotionsmittel: Petroleumether: Ethylacetat, 99 : 1) gereinigt und man erhielt 1C4 (84 mg, 89,14%).
1H NMR (CDCl3, 300 MHz) δH 1.52 (3H, s, CH 3), 2.14 und 2.21 (2H, jew. m, CHCH 2CH2), 2.80 and 3.26 (2H, 2 × d, J = 15.5 Hz, CCH 2), 3.04 und 3.13 (2H, 2 × d, J = 15.5 Hz, CCH 2), 3.11 (2H, m, CHCH2CH 2), 3.49 (1H, m, CHCH2CH2), 7.26 (8H, br m, 8 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 22.6 (CH3), 28.6, 31.9, 46.3, 46.5 (CH2), 55.5 (CH), 124.5, 124.7, 125.2, 125.8, 125.9, 125.9, 126.4, (Ar-CH), 142.5, 143.0, 145.1, 145.2 (Ar-C).
Referenzen: C M Wong, D Popies, R Schwerk und J Te Raa. Can, J. Chem. Bd. 49 (1971), 2714 - Reduktion mit Natriumboranat Synthese von 1C5
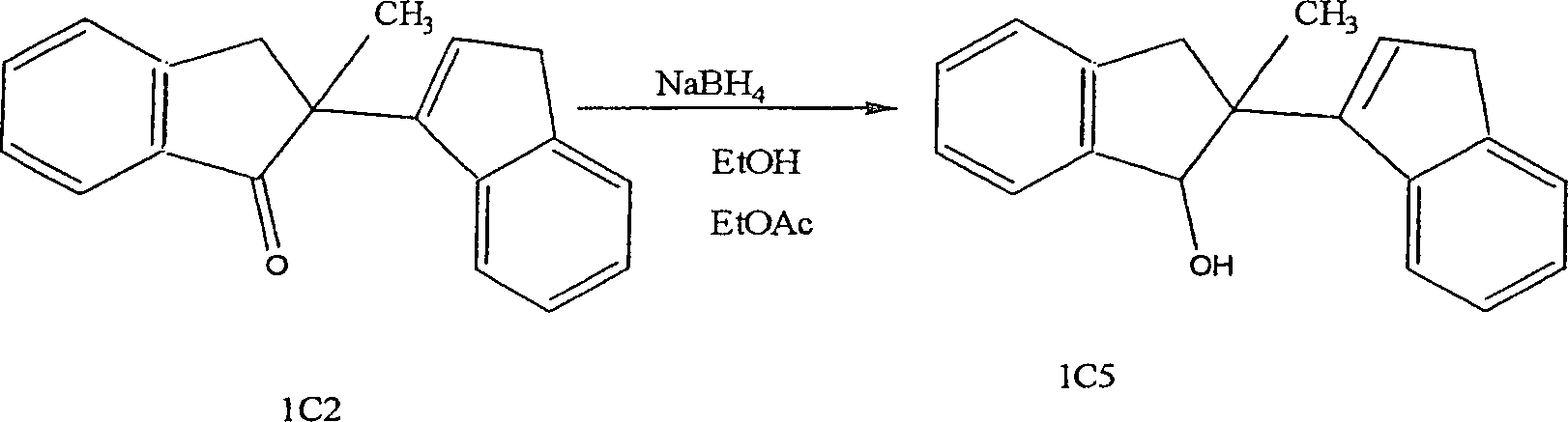
- 1C2 (530 mg, 2,04 mMol) wurde in Ethanol (10 ml) gelöst, und Natriumboranat (0,1 g, 2,63 mMol) wurde in die Reaktion in kleinen Portionen über 10 Minuten gegeben. Die Reaktion wurde bei Raumtemperatur für 3 Stunden gerührt. Das Reaktionsgemisch wurde in Wasser (20 ml) gegossen und in Diethylether (3 × 20 ml) extrahiert. Die Blitzsäulenchromatographie über Silikagel (Elutionsmittel: Petroleumether (Siedepunkt 40–60°C) : Ethylacetat, 98 : 2) ergab das Produkt 1C5 als klares Öl, 396 mg, 74,15%. Es wurde festgestellt, dass das Produkt als Mischung aus Diastereomeren erhalten wurde.
IR (KBr)max: 3429,8 cm–1 - Wo zu unterscheiden war, sind die Werte für das kleinere Diastereomere kursiv angegeben.
1H NMR (CDCl3, MHz) δH 1.44, 1.47 (3H, d, CH 3), 3.00 & 3.84, (1H, 2 × d, J = 15.5 Hz, CH of CH2), 3.13 & 3.35 (1H, dd, J = 15.9 Hz, CH of CH2), 3.43 & 3. 59 (2H, 2 × d, J = 2 Hz, 2 Hz, CH 2), 5.41, 5.67 (1H, 2 × s, CHOH), 6.49, 6.53, (1H, 2 × t, J = 2 Hz, 2 Hz, C=CH), 7.40 & 7.78 (6H, m & d, 6 × Ar-H), 7.61 (2H, m, 2 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 25.5 (CH3), 37.6, 37.9, 43.1, 43.2 (CH2), 49.9, 50.2 (qC), 80.7, 81.6 (CHOH), 121.1 (CH), 121.2, 121.6, 124.2, 124.3, 124.4, 124.5, 124.8, 125.1, 125.3, 125.8, 126.1, 126.4, 128.0, 128.6, 128.9, 128.9, 130.8, 138.5, 140.3, 142.8, 143.3, 143.9, 144.0, 145.4, 145.5, 148.2, 150.0, 171.3, (Ar-CH & Ar-C & 1 × C=CH). - Hydrazinhydratreaktion Modifizierte Reduktionsreaktion nach Huang-Minlon Synthese von 1C7

- 1C2 (100 mg, 0,38 mMol) wurde in Ethylglykol (5 ml) dispergiert. Es wurde Hydrazinhydrat (2,5 ml) zusammen mit Natriumhydroxid (0,2 g) hinzugegeben. Die Reaktion wurde unter Rückfluss für 24 Stunden gerührt. Das Reaktionsgemisch wurde auf Raumtemperatur abgekühlt, und es wurde Wasser (50 ml) hinzugegeben, und das Produkt wurde mit Ethylacetat (3 × 20 ml) extrahiert. Die organische Schicht wurde isoliert und über wasserfreiem Natriumsulfat getrocknet. Die Blitzsäulenchromatographie (Elutionsmittel : Petroleumether : Ethylacetat 99,1) ergab das reine Produkt 1C7, 34 mg, 35,58%.
Massenspektrum geringer Auflösung: gefunden: M+246
erforderlich: M+246
1H NMR (CDCl3, 300 MHz) δH 1.53 (3H, s, CH 3), 3.04 (2H, d, J = 15.5 Hz, CH 2), 3.39 (2H, s, CH 2), 3.59 (2H, d, J = 15.5 Hz, CH 2), 6.36 (1H, t CH), 7.28 (6H, br m, 6 × Ar-H), 7.48 (1H, m 1 × Ar-H), 7.53 (1H, br d, 1 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 27.5 (CH3), 37.4, 45.6, 45.6 (3 × CH2) 44.2 (qC), 121.3, 124.1, 124.2, 124.9, 124.9, 125.7, 126.3, 126.3, 126.9, (8 × Ar-CH und 1 × C-CH) 142.4, 142.4, 144.0, 145.4, 151.7 (4 × Ar-C und 1 × C=CH). - Synthese von 1C7 Reduktionsreaktion mit Cyanborhydrid

- 1C2 (100 mg, 0,38 mMol) wurden in 1,2-Dichlorethan (5 ml) bei Raumtemperatur dispergiert. In diese Lösung wurde festes Zinkjodid (0,02 g, 0,625 mMol) und Natriumcyanborhydrid (0,2 g, 3,18 mMol) gegeben. Die Reaktion wurde unter Rückfluss für 20 Stunden gerührt. Das Produkt wurde in Wasser (15 ml) gegeben und in Ethylacetat extrahiert. Es wurde die Blitzsäulenchromatographie (Elutionsmittel: Petroleumether: Ethylacetat 9 : 1) angewendet, um das 1C7, 27 mg, 29,13% zu isolieren.
1H NMR (CDCl3, 300 MHz) δH 1.53 (3H, s, CH 3), 3.04 (2H, d, J = 15.5 Hz, CH 2) 3.39 (2H, s, CH 2), 3.59 (2H, s, J = 15.5 Hz, CH 2) 6.36 (1H, t. J = 2.1 Hz, CH) 7.28 (6H, br m, 6 × Ar-H), 7.48 (1H, m, 1 × Ar-H), 7.53 (1H, br d, 1 × Ar-H).
Referenzen: C K Lau, Claude Durfresne, P C Belanger, S
Pietre und J Scheigetz
J Org Chem (1986), 51, 3038–3043 - Synthese von 1C8

- 1C5 (100 mg, 0,38 mMol) wurde in sauberem, trockenen DCM (5 ml) gelöst. In diese Lösung wurde Triethylamin (0,2 ml), DMAP (0,1 g) Essigsäureanhydrid (0,35 ml, 10 Äquivalente) gegeben. Das Reaktionsgemisch wurde bei Raumtemperatur für 15 Minuten gerührt und über eine Silikasäule gelassen, wobei mit Lösungsbenzin (Siedepunkt 40–60°C) : Ethylacetat (8 : 2) eluiert wurde, um das 1C8 (67 mg, 57,7%) zu erhalten.
1H NMR (CDCl3, 300 MHz) δH 1.44 (3H, s, CH 3), 1.53 (3H, s, OCOCH 3), 2.91– 3.91 (4H, br. m, CH 2), 6.38 (1H, br s, C=CH), 6.445 (1H, s, CHOCOCH3), 7.18– 7.58 (8H, m, Ar-H)
13C NMR (CDCl3, 75.47 MHz) δC 20.7, 21.1 (CH3), 26.1 (OCOCH3), 37.5, 37.6, 43.7, 43.9, (2 × CH2), 47.9, 48.7, (qC), 81.5, 82.4 (CHOCOCH3), 121.0, 121.3, 123.8, 124.1, 124.2, 124.4, 124.8, 125.1, 125.2, 125.8, 125.9, 126.7, 126.8, 127.2, 128.4, 128.6, 129.1, 129.3, 140.5, 141.8, 143.5, 143.8, 144.8, 145.3, 148.3 (8 × Ar-CH, 4 × Ar-C, 1 jew. C=CH), 170.6 (OCOCH3) - Synthese von 1C9

- In eine Lösung aus Indan-1-on (2 g, 0,015 Mol) in Diethylether (40 ml) und tert.-Butanol (20 ml) wurde unter Rühren Kaliumtert.-butoxid (1,7 g, 15,2 mMol) in Portionen gegeben. Nach der Zugabe des Kalium-tert.-butoxids wurde Jodmethan (2,13 g, 0,934 ml, 14,6 mMol) tropfenweise hinzugefügt, und die Reaktion wurde unter Rückfluss für 2 Stunden gerührt. Die Reaktion wurde auf Raumtemperatur abgekühlt und in Ethylacetat extrahiert. Das Lösungsmittel wurde bei reduziertem Druck verdampft. Die Säulenchromatographie über Silikagel mit Lösungsbenzin als Elutionsmittel (Siedepunkt 40–60°C) : Ethylacetat (9 : 1) ergab das 1C9 (0, 87 g, 39,9%).
1H NMR (CDCl3, 300 MHz) δH 1.31, 1.34, 1.61 (9H, 3 × s, 3 × CH 3), 3.20 (1H, d, J = 17.3 Hz, CH of CCH 2), 3.67 (1H, d, J = 17.3 Hz, CH of CCH 2), 6.31 (1H, s, C=CH, 6.79 (1H, d, J = 6.6 Hz, 1 × Ar-H), 6.80–7.17 (2H, m, 2 × Ar-H), 7.23– 7.31 (1H, t J = 72 Hz, 1 × Ar-H), 7.41–7.49 (2H, m, 2 × Ar-H), 7.62–7.70 (1H, t, J = 7.3 Hz, 1 × Ar-H), 7.87 (1H, d, J = 6.5 Hz, 1 × Ar-H)
13C NMR (CDCl3, 75.47 MHz) δC 23.9, 24.7, 24.8 (3 × CH3), 41.2 (1 × CH2), 47.8, 50.0 (2 × qC), 120.2, 121.4, 124.7, 125.1, 126.1, 126.7, 127.7, 135.1, 142.9 (8 × Ar-CH und 1 × C=CH), 135.7, 141.0, 141.6, 152.2, 154.5 (4 × Ar-C & 1 × C=CH), 208.2 (C=O) - Elementarmikroanalyse
- C21H20O erforderert C: 87,5, H: 6,94, gefunden C: 87,21, H: 7,07
- Synthese von 1C10

- Die Reaktionsausbeute von 1C10 betrug 0,87 g, 77,89%.
- Massenspektrum geringer Auflösung: gefunden: M+274, M+ – 29 = 245
erforderlich: M+274
1H NMR (CDCl3, 300 MHz) δH 0.83 (3H, t, J 8 Hz, CH3), 2.21 (2H, m, J = 8 Hz, CH2), 3.34 (1H, d, CH), 3.35 (2H, s, CH2), 3.59 (1H, d, J = 17 Hz, CH) 6.49 (1H, t, J = 2 Hz, CH), 7.15–8.0 (8H, m, 8 × Ar-H)
13C NMR (CDCl3, 75.47 MHz) δC 8.6 (CH2CH3), 29.5, 37.4, 38.3 (3 × CH2), 54.4, (qC), 120.1, 123.9, 123.9, 124.4, 125.7, 126.2, 127.3, 129.7, 134.8, (8 × Ar-CH & C=CH), 136.9, 143.2, 144.8, 145.1, 152.6, (5 × Ar-C), 207.7 (C=0) - Synthese von 1C12

- Die Reaktionsausbeute für 1Cl2 betrug 0,78 g, 67,09%.
- Massenspektrum geringer Auflösung: gefunden: M+286
erforderlich: M+286
1H NMR (CDCl3, 300 MHz) δH 2.94 (2H, d, CH 2CH=CH2), 3.38 (2H, br s, C=CHCH 2), 3.53 (2H, ab q, J = 17.5 Hz, CH 2), 4.99 (1H, dd, J = 1 Hz, 10 Hz, CH2CH=CH 2), 5.16 (1H, dd, J = 3.3 Hz, 17 Hz, CH2CH=CH 2), 5.62 (1H, m, CH2CH=CH 2), 6.52 (1H, t, J = 2 Hz, C=CHCH2), 7.06 (1H, m, 1 × Ar-H), 7.18 (2H, m, 2 × Ar-H), 7.46 (3H, m, 3 × Ar-H), 7.65 (1H, dt, J = 1.3 Hz & J = 7.6 Hz 1 × Ar-H), 7.87 (1H, d, J = 7.5 Hz, 1 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 37.6, 37.6, 41.1 (3 × CH2), 53.9 (qC), 118.7 (CH2C=CH2), 120.2, 124.0, 124.2, 124.6, 125.8, 126.4, 127.6,130.3, 132.9, 135.2, (8 × Ar-CH & 2 × CH), 136.2, 143.0, 144.8, 144.9, 152.7 (Ar-C) , 207.4 (C=O) - Wilkinson-Reduktion Synthese von 1C13

- 1C12 (100 mg, 0,349 mMol) wurde in Ethanol (20 ml) und Ethylacetat (10 ml) gelöst. In diese Lösung wurde der Wilkinson-Katalysator (0,1 g) unter Rühren hinzugegeben. Die Reaktion wurde dann unter Wasserstoff für 20 Stunden gerührt. Das Produkt war zwischen dem Ethylacetat und Wasser aufgeteilt, und die organische Schicht wurde isoliert und mit Na2SO4 getrock net. Das Rohprodukt wurde über die Blitzsäulenchromatographie mit einer Ausbeute von 1C13 mit 57 mg, 56,60% gereinigt.
1H NMR (CDCl3, 300 MHz) δH 0.88 (3H, t, J = 7 Hz, CH 3), 1.27 (2H, m, CH 2), 2.16 (2H, m, CH 3), 3.36 (2H, br.s, C=CHCH 2), 3.49 (2H, ab g, J = 17.6 Hz, COCH 2) 6.50 (1H, t, J = 2 Hz, CH), 7.12 (3H, m, 3 × Ar-H), 7.50 (3H, m, 3 × Ar-H), 7.64 (1H, dt, J = 1.2 Hz & J = 7.6 Hz, 1 × Ar-H), 7. 86 (1H, d, J = 7.2 Hz, 1 × Ar-H)
13C NMR (CDCl3, 75.47 MHz) δC 14.5 (CH3), 17.6, 37.6, 38.9, 39.2 (4 × CH2), 54.3 (qC), 120.3 (CH), 124.1, 124.2, 124.6, 125.8, 126.3, 127.6, 129.8, 135.0, (8 × Ar-CH), 136.9, 143.3, 144.9, 145.3, 152.8 (4 × Ar-C & 1 × C=C), 208.2 (C=O). - Synthese von 1C15 Natriumborhydridreduktion

- Die Methode ist die gleiche wie die, die für 1C5 beschrieben wurde.
- Die Reaktionsausbeute für 1C15 war (73 mg, 72,42%), aus der zwei Diastereomere über die Blitzsäulenchromatographie getrennt wurden.
- Diastereomeren-Mischung 1:
- 1H NMR (CDCl3, 300 MHz) δH 2.55 & 2.61 (1H, 2 × d, CCH 2CH=CH2), 2.89 & 2.94 (1H, 2 × d J = 6.15 Hz CCH 2CH=CH2), 3.33 (2H, g, J = 16 Hz, CH 2), 3.35 (2H, d, J = 2 Hz, C=CHCH 2), 4.90 (1H, dd, J = 1 Hz, 10 Hz, CH2CH=CH 2), 4.98 (1H, dd, J = 3.3 Hz, 17 Hz, CH2CH=CH 2), 5.51 (1H, d, J = 7.9 Hz, CHOH), 5.64 (1H, m, C=CHCH2), 6.40 (1H, s, C=CH) 7.19 (5H, m, 5 × Ar-H), 7.35 (1H, m, 1 × Ar-H), 7.50 (1H, d, J = 5 Hz, 1 × Ar-H), 7.72 (1H, d, J = 5 Hz, 1 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 37.4, 37.6, 40.4 (3 × CH2), 53.0 (qC), 81.8 CHOH), 116.9 (CH=CH2), 121.7, 124.1, 124.1, 124.4, 124.7, 125.8, 126.9, 128.4, 129.9, 136.1, (8 × Ar-CH & 1 × C=CH & 1 × CH=CH2), 140.7, 143.9, 144.1, 45.3, 148.2 (4 × Ar-C & C=CH). - Diastereomeren-Mischung 2:
- 1H NMR (CDCl3, 300 MHz) δH 2.24 (1H, d, J = 5.39 Hz, CHOH), 2.87–3.41 (6H, m, 3 × CH2), 4.91 (2H, m, CH=CH2), 5.49 (H, d, J = 5.37 Hz, CHOH), 5.62 (1H, m, CH=CH2), 6.37 (1H, s, C=CHCH2), 7.23–7.72 (8H, m, 8 × Ar-CH).
13C NMR (CDCl3, 75.47 MHz) δC 29.7, 37.4, 37.6 (3 × CH2), 81.7 COH), 116.9 (CH=CH2), 121.7, 124.0, 124.1, 124.4, 124.7, 125.8, 126.9, 128.3, 129.9, 136.1, (8 × ArCH & 2 × C=CH), 140.7, 143.8, 144.0, 145.3, 148.2 (5 × Ar-C). - Synthese der Acetate Synthese von 1C16

- 1C15 (140 mg, 0,5 mMol) wurde in sauberem, trockenen DCM (10 ml) gelöst. In diese Lösung wurde Triethylamin (0,15 g, 0,20 ml) DMAP (0,1 g) und Essigsäureanhydrid (0,45 ml, 10 Äquivalente) gegeben. Das Reaktionsgemisch wurde bei Raumtemperatur für 15 Minuten gerührt und durch eine Silikasäule mit Lösungsbenzin (Siedepunkt 40–60°C): Ethylacetat (8 : 2) gelassen und man erhielt 1C16 (149 mg, 92,9%).
1H NMR (CDCl3, 300 MHz) δH 1.53 (3H, s, OCOCH 3), 2.23 (1H, dd, J = 5.5 und 13.8 Hz, CH of CH 2), 3.22 (1H, d, J = 15.8 Hz, CH of CH 2), 3.44 (2H, s, CH 2), 3.63 (1H, d, J = 15.8 Hz, CH of CH 2), 4.87 (1H, d, J = 17 Hz, CH of CH 2), 4.95 (1H, d, J = 10 Hz, CH of CH 2), 5.61–5.49 (1H, m CH), 6.36 (1H, t, J = 2.0 Hz, C=CH, 6.54 (1H, s, CHOCOCH3), 7.21–7.37 (5H, m, 5 × Ar-H), 7.51 (2H, t, J = 6.6 Hz, 2 × Ar-H), 7.63 (1H, d, J = 7.2 Hz, 1 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 20.6 (OCOCH3), 37.6, 40.0, 40.3 (3× CH2), 51.6 (qC), 81.7 (CHOCOCH3), 117.8 (CH=CH2), 120.8, 123.8, 124.2, 124.9, 125.9, 126.8, 127.1, 129.3, 130.9, 134.1, 140.7, 143.1, 143.6, 144.7, 145.9, (8 × Ar-CH, 4 × Ar-C, 1 jew. C=CH und 1 × CH=CH2), 170.6 (OCOCH3). Synthese von 1C17 & 1C18 Natriumborhydrid-Reduktion von 1C13Ausbeute (90 mg, 90%)
- 1C17
- 1H NMR (CDCl3, 300 MHz) δH 0.84–2.20 (8H, br m, CH 3, CH2's), 3.29 (2H, s, C=CHCH 2), 3.33 (2H, abq, J = 16 Hz, COHCCH 2), 5.42 (1H, br s, CHOH), 6.28 (1H, t, J = 2.1 Hz, C=CH), 7.18–7.81 (8H, m, 8 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δH 14.8 (CH3), 18.7 34.7, 37.5, 40.8, (4 × CH2), 53.6 (qC), 81.2 (CHOH), 121.7, 124.0, 124.3, 124.4, 124.7, 125.8, 126.7, 128.3, 129.7, 141.7, 144.1, 144.2, 145.3, 148.2, (8 × Ar-CH & 4 × Ar-C & 1 × C=CH & 1 × C=CH). - 1C18
- 1H NMR (CDCl3, 300 MHz) δH 0.74 (3H, t, J = 7.4 Hz, CH2CH2CH 3), 0.92 (2H, m, CCH2CH 2CH3), 1.75 (2H, m, CCH 2CH2CH3), 3.45 (2H, s, CCH 2), 3.12 & 3.59 (1H jew. d. J = 15.6 Hz, CHCH 2), 5.37 (1H, s, CHOH), 6.45 (1H, s, C=CH), 7.27– 7.79 (8H, m, 8 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 14.4 (CH 3), 18.5, 29.7, 37.8, 39.4, (4 × CH 2), 54.8 (qC), 81.8 (CHOH), 120.9, 124.2, 124.7, 125.1, 125.7, 126.3, 126.7, 128.7, 130.9, 142.6, 143.4, 143.7, 145.2, 146.1, (8 × Ar-CH & 4 × Ar-C & a × C=CH & 1 × C=CH). - Synthese von 1C19

- 1C18 (70 mg, 0,25 mMol) wurde in sauberem, trockenen DCM (5 ml) gelöst. In diese Lösung wurden Triethylamin (15 ml), DMAP (0,5 g) und Essigsäureanhydrid (0,25 ml, 10 Äquivalente) hinzugefügt. Das Reaktionsgemisch wurde bei Raumtemperatur für 15 Minuten gerührt und durch eine Silikasäule mit Lösungsmittel benzin (Siedepunkt 40–60°C) : Ethylacetat (8 : 2) gelassen, um das 1C19 (65 mg, 81,1%) zu erhalten.
1H NMR (CDCl3, 300 MHz) δH 0.71 (3H, t, J = 7.1 Hz, CH2CH 3), 0.89–1.97 (4H, br. m, CH 2's), 1.51 (3H, s, CH 3), 3.17 (1H, d, J = 15.5 Hz CH of CHCH 2), 3.40 (2H, s, CH 2), 3.62 (1H, d, J = 15.6 Hz, CH v. CH 2), 6.34 (1H, t, J = 2.2 Hz, C=CH), 6.49 (1H, s, CHOCOCH3), 7.18–7.59 (8H, m, Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 14.3 (CH2 CH3), 18.3 (CH2), 20.7 (OCOCH3) 37.6, 38.6, 40.4 (3 × CH2), 52.4, (qC), 82.4 (CHOCOCH3) 120.8, 123.8, 124.2, 124.98, 125.9, 126.8, 127.1, 129.3, 130.9, (8 × Ar-CH, Vinyl-CH), 141.0, 143.4, 143.9, 144.6, 146.6, (4 × Ar-C, und 1 × C=CHCH2), 170.7 (OCOCH3). - Synthese von 1C20
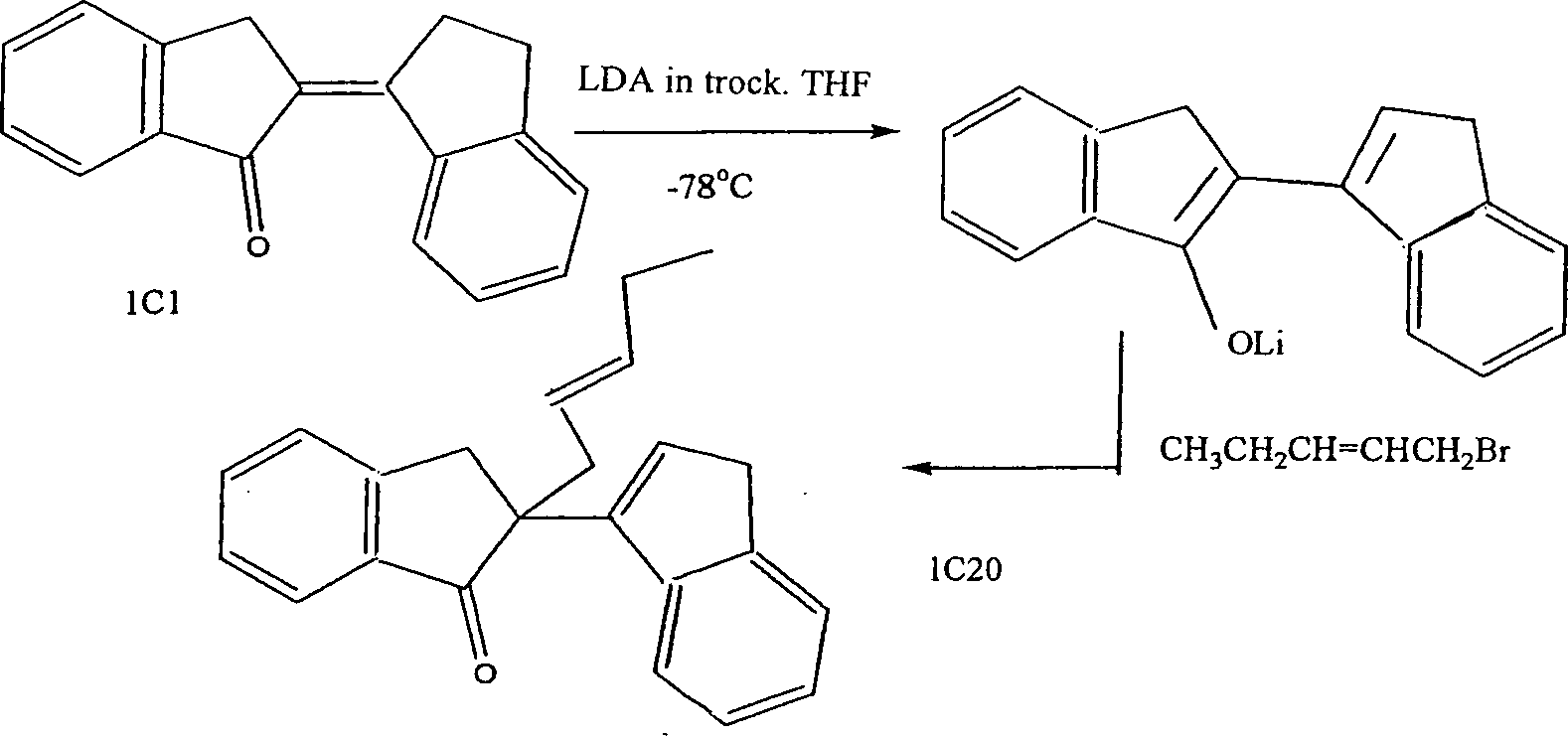
- Die Reaktion für 1C20 war 40%.
1H NMR (CDCl3, 300 MHz) δH 0.73 (3H, t, CH2CH 3), 1.83 (2H, m, CH 2CH3), 2.85 (2H, d, CH 2CH=CH), 3.8 (2H, br s, C=CHCH 2), 3.50 (2H, ab g, J = 13.0 Hz, COCCH 2), 5.18 & 5.58 (2H, 2 × m, CH2CH=CHCH2), 6.52 (1H, t, J = 2 Hz, 1 × C-CHCH2), 7.01 (1H, m, 1 × Ar-H), 7.15 (2H, m, 2 × Ar-H), 7.40 (3H, m, 3 × Ar-H), 7.65 (1H, t, 1 × Ar-H), 7.85 (1H, d, 1 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δH 13.4 (CH3), 25.4, 37.6, 37.6, 39.9 (4 × CH2), 54.3 (qC), 120.1, 123.1, 124.0, 124.0, 124.6, 125.8,126.3, 127.4, 130.2, 135.0, 136.6, 136.9, 143.2, 144.9, 145.1, 152.9, (8 × Ar-CH & 4 × Ar-C & 1 × C=CH & 1 × CH=CH), 2078.9 (C=O). - Synthese von 1C21 Wilkinson-Reduktion von 1C20

- 1C20 (0,50 g, 1,58 mMol) wurde in Ethanol (20 ml) und Ethylacetat (10 ml) gelöst. In diese Lösung wurde der Wilkinson-Katalysator (0,1 g) unter Rühren hinzugegeben. Die Reaktion wurde dann unter Wasserstoff für 20 Stunden gerührt. Das Produkt war zwischen dem Ethylacetat und Wasser aufgeteilt, und die organische Schicht wurde isoliert und Na2SO4 getrocknet.
- Das Rohprodukt wurde über die Blitzsäulenchromatographie gereinigt, und man erhielt das 1C21 (450 mg, 90%).
1H NMR (CDCl3, 300 MHz) δH 0.86 (3H, t, J = 1.6 Hz, CH 3), 1.27-1.47 (6H, m, 3 × CH 2), 2.16–2.21 (2H, m, CH 2), 3.37 (2H, s, CH 2), 3.52 (2H, ab q, J = 17.6Hz, CHCH2), 6.51 (1H, d, J = 2.1 Hz, CH), 7.15–7.91 (8H, m, 8 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 13.9 (CH3), 22.3, 23.8, 32.2, 36.8, 37.6, 38.8 (6 × CH2), 54.3 (qC), 120.3, 124.0, 124.1, 124.5, 125.8, 126.3, 127.3, 127.5, 129.8, 134.9 (8 × Ar-CH & 1 × C=CH), 136.8, 143.3, 144.9, 145.3, 152.7 (4 × Ar-C & 1 × C=CH), 208.1 (C=O). - Synthese von 1C22 & 1C23 Natriumborhydrid-Reduktion von 1C20

- 1020 (0,50 g, 1,58 mMol) wurde in Ethanol und Ethylacetat (2 : 1, 9 ml) gelöst, und es wurde Natriumborhydrid (0,1 g, 0,263 mMol) in die Reaktion in kleinen Portionen über 10 Minuten gegeben. Die Reaktion wurde bei Raumtemperatur für 3 Stun den gerührt. Die Reaktionsmischung wurde auf Wasser (20 ml) gegossen und in Diethylether (3 × 20 ml) extrahiert. Die Blitzsäulenchromatographie über Silikagel mit dem Elutionsmittel: Petroleumether (Siedepunkt 40–60°C) : Ethylacetat, 98 : 2) ergab 1C22 & 1C23 (470 mg, 95%).
- 1C22
- 1H NMR (CDCl3, 300 MHz) δH 0.79 (3H, t, J = 7.4 Hz, CH2CH 3), 1.83–3.369 (8H, br m, CH2's), 5.21–5.53 (2H, m, CH2CH=CHCH2), 5.54 (1H, br s, CHOH), 6.21, 6.43 (1H, 2 × s, CH=C), 7.21–7.71 (8H, m, 8 × Ar-H).
- 1C23
- 1H NMR (CDCl3, 300 MHz) δH 0.34–3.52 (12H, m, CH2's), 5.09–5.29 (2H, m, CH=CH), 5.36 (1H, br. m, CHOH), 6.42 (1H, d, J = 6.5 Hz, CH=C), 7.23–7.76 (8H, m, 8 × Ar-H)
- Synthese von 1024

- Die Reaktionsausbeute für 1C24 betrug 230 mg, 33,67%.
Massenspektrum geringer Auflösung: gefunden: M+336 M+ – 91 + 245
erforderlich: M+336
1H NMR (CDCl3, 300 MHz) δH 3.37 (2H, dd, C=CHCH 2), 3.55 (2H, ab g, J = 13 Hz, CCH 2), 3.54 (2H, d, J = 14 Hz, PhCH 2), 6.53 (1H, t, J = 2 Hz, C=CH), 7.12 (5H, m, 5 × Ar-H), 7.25 (5H, br m, 5 × Ar-H), 7.47 (2H, m, 2 × Ar-H), 7.78 (1H, d, J = 7Hz, 1 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 36.8, 37.6, 41.7 (3 × CH2), 55.5 (qC), 120.5, 123.9, 124.2, 124.7, 125.9, 126.1, 126.4, 127.7, 127.2, 130.2, 130.2, 130.4, 134.8, (13 × Ar-CH & 1 × C=CH), 136.6, 136.7, 143.1, 145.1, 145.1, 152.6 (5 × Ar-C & C=CH). - Natriumborhydrid-Reduktion Synthese von 1C25 & 1C26

- 1C25 und 1C26 wurden in einer Ausbeute von 90 mg, 89,45% als Diastereomerengemisch isoliert.
1H NMR (CDCl3, 300 MHz) δH 2.58–3.62 (6H, br m, 3 × CH 2), 5.45 & 5.56 (1H, 2 × br s, CHOH), 6.04 & 6.09 (1H, 2 × s, C=CHCH2), 6.64 (1H, d, J = 2 Hz, Ar-CH), 7.15 (9H, m, 9 × Ar-CH), 7.67 (1H, d, Ar-CH), 7.98 (1H, dd, Ar-CH), 7.90 & 8.10 (1H, 2 × d, J = Hz, 1 × Ar-CH).
13C NMR (CDCl3, 75.47 MHz) δC 37.1, 37.4, 37.8, 38.4, 39.6, 40.9 (5 × CH2), 55.2, 55.8, (qC), 81.2, 81.3 (CHOH), 120.9, 122.3, 123.9, 124.3, 124.4, 124.5, 124.7, 124.7, 124.9, 125.1, 125.8, 125.9, 126.1, 126.1, 126.5, 126.5, 126.7, 126.9, 127.3, 127.4, 127.4, 128.4, 129.0, 130.2, 130.2, 130.2, 131.2, 134.2, (Ar-CH und C=CH), 137.8, 138.7, 141.2, 142.4, 143.2, 143.6, 144.2, 145.2, 145.2, 147.4 (Ar-C). - Synthese von 1C27
- 1C25/1C25 (50 mg, 1,5 mMol) wurden in sauberem, trockenen DCM (5 ml) gelöst. In diese
-

- Lösung wurden Triethylamin (0,1 mlg), DMAP (0,05 g) und Essigsäureanhydrid (0,25 ml, 10 Äquivalente) gegeben. Die Reaktionsmischung wurde bei Raumtemperatur für 15 Minuten gerührt und durch eine Silikasäule mit Lösungsmittelbenzin (Siedepunkt 40–60°C) : Ethylacetat (8 : 2) als Elutionsmittel gelassen, wobei man das 1C27 (48 mg, 85,4%) als Diastereomerenmischung erhielt.
1H NMR (CDCl3, 300 MHz) δH 1.56 und 2.23 (6H, 2 × s, 2 × OCOCH 3), 2.57– 3.73 (12H, m, 6 × CH 2), 5.59 und 6.6 (4H, 2 × m, 2 × CH, 2 × CHCOCH3), 6.90– 7.73 (25H, m, 25 × Ar-H), 8.1 (1H, d, J = 6 Hz, 1 × Ar-H)
13C NMR (CDCl3, 75.47 MHz) δC 20.8, 21.4 (OCOCH3), 37.4, 37.5, 38.1, 39.2, 40.6, 41.0, (3 × CH2), 53.4, 54.1, (qC), 81.2, 81.7 (CHOCOCH3), 120.6, 121.9, 124.0, 124.1, 124.3, 124.4, 124.9, 126.0, 126.1, 126.2, 126.6, 126.9, 127.3, 127.5, 129.0, 129.5, 129.9, 130.5, 132.2, 132.24, 137.2, 138.3, 140.8, 143.2, 143.4, 143.5, 143.8, 144.6, 144.9, 145.2 (Ar-CH, Vinyl-C und Ar-C), 170.5 (OCOCH3). - Synthese von 1C31

- Die Reaktionsausbeute für 1C31 betrug 60%.
1H NMR (CDCl3, 300 MHz) δH 3.37 (2H, dd, J = 1.8 Hz, C=CHCH 2), 3.45, 3.56, (2H, d, PhCH 2), 3.57 (2H, q, J = 13.0 Hz, C-CH 2), 3.84, (3H, s, CH3), 6.48 (1H, t, J = 1.8 Hz, CH), 7.25 (7H, m, 7 × Ar-H), 7.46 (2H, dt, 2 × Ar-H), 7.77 (3H, m, 3 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 36.9, 37.6, 41.6 (3 × CH2), 51.9 (CH3), 55.4 (qC), 128.4 (Ar-C), 120.5, 124.0, 124.3, 124.8, 125.9, 126.1, 127.5, 129.2, 129.2, 130.2, 130.2, 130.6, 135.1, (12 × Ar-CH & 1 × CH=C), 136.5, 142.1, 142.9, 144.6, 145.1, 152.3, (5 × Ar-C & 1 × C=CH), 166.8 (CO2CH3), 207.2 (C=O). - Hydrolyse von 1C31 Synthese von 1C32

- Der Benzoatester 1C31 (0,1 g, 0,253 mMol) wurde in einer Lösung aus 1,45 Mol NaOH in THF-MeOH-H2O (6 : 3 : 2) (4 ml) gelöst, und dann unter Rückfluss gehalten. Nach 20 Minuten zeigte die TLC, dass die Hydrolyse des Benzoatesters 1031 vollständig war. Nach dem Abkühlen der Reaktionsmischung, fügte man eine gesättigte Lösung aus wässrigem Ammoniumchlorid (4 ml), wässriger HCl (2 Mol) (10 ml) und Ether (30 ml) hinzu. Die organische Schicht wurde isoliert, und die wässrige Schicht wurde mit Ether (1 × 10 ml) extrahiert. Die vereinten organischen Extrakte wurden mit Na2SO4 getrocknet und filtriert. Nach der Verdampfung blieb die Säure 1C32 als leicht gefärbter Feststoff zurück.
1H NMR (CDCl3, 300 MHz) δH 3.39 & 3.45 (2H, dd, J = Hz C=CHCH 2), 3.49 & 3.57 (2H, d, PhCH 2), 3.59 (2H, q, C-CH 2), 6.49 (1H, br s, CH, 7.22 (8H, m 8 × Ar-H), 7.47 (2H, t, 2 × Ar-H), 7.79 (1H, d, 1 × Ar-H), 7.89 (2H, d, 2 × Ar-H)
13C NMR (CDCl3, 75.47 MHz) δC 36.9, 37.7, 41.7 (3 × CH2), 55.4, (qC), 120.5, 124.1, 124.3, 124.8, 126.0, 126.2, 127.4, 129.8, 129.8, 130.3, 130.3, 130.7, 136.4 (12 × Ar-CH & 1 × C=CH), 135.2, 135.2, 142.9, 143.1, 144.6, 145.1, 152.3 (6 × Ar-C & 1 × C=CH), 171.6 (CO2H), 207.3 (C=O). - Synthese von 1C33

- Die Reaktionsausbeute für 1C33 betrug 0,53 g, 41,12%.
1H NMR (CDCl3, 300 MHz) δH 3.31 (2H, q, J = 16.2 Hz, COCH 2), 3.30 (2H, dd, J = 2 Hz, C=CHCH2), 3.54 (3H, s, COOCH 3), 3.65 (2H, ab q, CH 2COOCH3), 6.31 (1H, t, CH), 7.25 (3H, m 3 × Ar-H), 7.42 (3H, m, 3 × Ar-H), 7.63 (1H, dt, 1 × Ar-H), 7.91 (1H, m 1 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 37.6, 38.2, 39.8 (3 × CH2), 51.6 (CH3), 52.0 (qC), 120.3, 124.3, 124.3, 124.8, 125.9, 126.3, 127.5, 130.4, (8 × Ar-CH), 134.9 (CH), 136.1, 142.5, 143.6, 145.0, 152.2 (5 c Ar-C & 1 × C=CH), 171.4 (CO2CH3), 206.2 (C=0). - Synthese von 1C34 Hydrolyse von 1C33

- 1C33 (0,1 g, 0,316 mMol) wurde in einer Lösung aus 1,45 Mol NaOH in THF : MeOH : H2O (6 : 3 : 2) (4 ml) gelöst, und dann unter Rückfluss gehalten. Nach 0,5 h zeigte die TLC, dass die Hydrolyse von BRA 64 vollständig war. Dieses wurde dann zwischen DCM (50 ml) und verdünnter HCl (1 Mol 20 ml) aufgeteilt. Die organische Schicht wurde isoliert und die wässrige Schicht mit DCM (2 × 50 ml) extrahiert. Die vereinten organischen Extrakte wurden mit Wasser gewaschen und über Na2SO4 getrocknet und filtriert. Nach der Verdampfung blieb die Säure 1C34 als Gummi zurück.
1H NMR (CDCl3, 300 MHz) δH 3.26 (4H, br m, C=CHCH2 & CH2COOH), 3.64 (2H, ab q J = 17.0 Hz, COCH 2), 6.28 (1H, t, J = 2 Hz, C=CHCH2), 7.18 (3H, br m, 3 × Ar=H), 7.45 (3H, br m, 3 × Ar-H), 7.65 (1H, dt, 1 × Ar-H). 7.86 (1H, br d, 1 × Ar-H). - Synthese von 1035

- Synthese von 1C38

- 1H NMR (CDCl3, 300 MHz) δH 1.88 (3H, s, OCOCH 3), 3.55 (2H, t, J = 2.7 Hz, C=CHCH2), 3. 58 (2H, ab q, J = 17.0 Hz, CCH2), 4.55, 4.91 (2H, 2 × d, J = 10.7 Hz, CH 2O-COCH3), 7.16 (3H, br m, 3 × AR-H), 7.45 (3H, m, 3 × AR-H), 7.65 (1H, dt, J = 6 Hz, 1.32 Hz 1 × Ar-H), 7.9 (1H, d, J = 6 Hx, 1 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 20.5 (CH3COO), 36.3, 378 (2 × CH2), 54.2 (CCO), 66.7 (CH2OCOCH3), 120.1, 124.1, 124.4, 142.6, 144.7, 152.6 (8 × Ar-CH & 4 × Ar-C & 1 × C=CH), 170.6 (CH3 COO), 205.1 (C=O). - Verknüpfungsreaktion des entsprechenden Silylenolethers von Indan-2-on an das entsprechende Dimethylacetal von Indan-2-on Synthese eines Silylenolethers von Indan-2-on

- In eine gerührte Lösung aus Indan-2-on (1,0 g, 7,57 mMol) und Triethylamin (0,84 g, 1,16 ml, 8,32 mMol) in Dichlormethan bei 0°C wurde Trimethylsilyltrifluormethansulfonat (1,68 g, 1,36 ml, 7,58 mMol) gegeben. Man ließ die Lösung bei 0°C für 15 Minuten rühren, und dann wurde die Lösung schnell durch eine Silikasäule mit Lösungsmittelbenzin (Siedepunkt 40–60°C) : Ethylacetat 100 : 0,5 als Elutionsmittel gelassen. Nach der Verdamp fung des Elutionsmittels wurde der Silylenolether als klares farbloses Öl (7), 0,50 g, 77,0% isoliert.
- Synthese eines Dimethylacetals von Indan-2-on

- In eine gerührte Lösung aus Indan-2-on (1,0 g, 7,57 mMol) in Methanol (12 ml) wurde Trimethylorthoformat (2 ml) und p-Toluolsulfonsäure (etwa 1 Mol-%) gegeben. Man ließ die Lösung dann bei Raumtemperatur für 2 Stunden rühren. In diese Lösung wurde dann festes Natriumbicarbonat (etwa 0,50 g) gegeben. Das Methanol wurde aus der Reaktionsmischung verdampft. Der Rohfeststoff wurde dann zwischen Ether : Wasser (1 : 1) (50 ml) verteilt. Die organische Schicht wurde isoliert und die wässrige Schicht mit Ether (3 × 20 ml) extrahiert. Die vereinten organischen Schichten wurden mit Natriumsulfat getrocknet. Nach der Verdampfung des Lösungsmittels ließ man dann das Rohprodukt durch eine Silikasäule mit Petroleumether zu 100% in der Aufteilung Petroleumether : Ethylacetat, 100 : 1 als Elutionsmittel. Nach der Verdampfung des Elutionsmittels wurde das Dimethylacetat von Indan-2-on als klares farbloses Öl, 0,80 g, 60 isoliert.
1H NMR (CDCl3, 300 MHz) δH 3.21 (4H, s, 2 × CH 2), 3.35 (6H, s, 2 × ⟐ OCH 3), 7.22 (4H, s, 4 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 41.2, 41.2 (2 × CH2), 49.4, 49.4, (2 × OCH3), 111.4 (C(OMe)2), 124.5, 124.5, 126.5, 126.5 (4 × Ar-CH), 139.8, 139.8 (2 × qC) - Synthese von 2C1

- In eine gerührte Lösung des Silylenolethers von Indan-2-on (7) (0,80 g, 3,92 mMol) und dem entsprechenden Dimethylacetal von Indan-2-on (0,70 g, 3,92 mMol) in Dichlormethan bei –78°C, wurde eine katalytische Menge von TMS-Triflate (30 μl) gegeben. Man ließ die Lösung bei –78°C für 3 Stunden stehen und ließ sie dann für 1 Stunde –50°C erreichen. In diese Lösung wurde dann eine 5%ige Lösung aus Natriumbicarbonat (etwa 20 ml) gegeben. Die organische Schicht wurde isoliert und die wässrige Schicht mit Dichlormethan (2 × 20 ml) extrahiert. Die vereinten organischen Schichten wurden mit Natriumsulfat getrocknet. Nach der Verdampfung des Lösungsmittels ließ man das Rohprodukt durch eine Silikasäule mit Petroleumether zu 100 in der Abstufung Petroleumether : Ethylacetat, 100 : 4, gehen. Nach der Verdampfung des Lösungsmittels wurde das 2C1 als Feststoff, 0,72 g, 74,3% isoliert.
Massenspektrum geringer Auflösung: gefunden M+246
erforderlich: M+246
1H NMR (CDCl3, 300 MHz) δH 3.45 (2H, s, CH 2), 4.10 (2H, s, CH 2), 4.40 (2H, s, CH 2, 7.45 (8H, m, 8 × AR-H)
13C NMR (CDCl3, 75.47 MHz) δC 40.7, 41.2, 42.7, (3 × CH2), 123.5, 124.3, 124.6, 125.1, 126.7, 127.0, 127.2, 127.5 (8 × Ar-CH), 129.6, 137.4, 139.2, 140.2, 141.1, 154.2 (4 × Ar-C & 2 × C=C), 204.0 (C=O). - Synthese von 2C1 Kalium-tert.-butoxid-Methode

- Kalium-tert.-butoxid (4,25 g, 37 mMol) in tButanol (125 ml) und Ether (10 ml) wurden tropfenweise über 20 Minuten in eine Lösung aus Indan-2-on (5,0 g, 37 mMol) in Ether (25 ml) und tButanol (5 ml) unter Rühren gegeben. Man ließ dann das Reaktionsgemisch über Nacht unter Rühren stehen.
- Das Rohprodukt verteilte sich zwischen dem Ethylacetat und dem gesättigten wässrigen Ammoniumchlorid. Die organische Schicht wurde isoliert und die wässrige Phase wurde mit Ethylacetat wieder extrahiert. Die organischen Schichten wurden vereint und über Natriumsulfat getrocknet. Nach der Verdampfung des Lösungsmittels erhielt das Rohprodukt. Es wurde die Blitzsäulenchromatographie angewendet, um das gewünschte Produkt zu reinigen (Elutionsmittel : Petroleumether (Siedepunkt 40-60°C): Ethylacetat, 9 : 1). Man erhielt nach der Umkristallisierung mit Ether das 2C1 als weißen kristallinen Feststoff, 0,41, 12%.
Massenspektrum geringer Auflösung: gefunden M+246
erforderlich: M+246
1H NMR (CDCl3, 300 MHz) δH 3.45 (2H, s, CH 2), 4.10 (2H, s, CH 2), 4.40 (2H, s, CH 2), 7.45 (8H, m, 8 × AR-H)
13C NMR (CDCl3, 75.47 MHz) δC 40.7, 41.2, 42.7, (3 × CH2), 123.5, 124.3, 124.6, 125.1, 126.7, 127.0, 127.2, 127.5 (8 × Ar-CH), 129.6, 137.4, 139.2, 140.2, 141.1, 154.2 (4 × Ar-C & 2 × C=C), 204.0 (C=O). - Synthese von 2C3

- Die Reaktionsausbeute für 2C3 betrug 27 mg, 24,24%.
1H NMR (CDCl3, 300 MHz) δH 0.71 (3H, t, CH2CH 3), 2.05 & 2.40 (2H, 2 × br m, CH 2CH3), 3.40 (2H, d, HC=CCH 2), 3.57 (2H, ab q, COCH 2), 6.34 (1H, br s, CH=C), 7.25 (8H, br m, 8 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 9.5 (CH2 CH3), 29.7, 38.4, 42.7 (3 × CH2), 62.5 (qC), 120.7, 123.6, 124.5, 124.8, 125.2, 126.3, 127.7, 127.7, 129.9 (8 × Ar-CH & 1 × C=CH), 136.7, 143.5, 143.9, 144.1, 129.7 (4 × Ar-C & 1 × C=CH), 216.1 (C=O). - Synthese von 2C4 und 2C5

- 2C1 (100 mg, 0,04 mMol) wurde in Ethanol (10 ml) und Ethylacetat (5 ml) dispergiert, und dieses wurde mit 5% Palladium auf Kohlenstoff (0,01 g) versetzt. Die Mischung wurde unter Stickstoff für 14 Stunden gerührt. Der Palladiumrückstand wurde durch Filtration entfernt, und das Lösungsmittel wurde unter Bildung des Rohreaktionsprodukts entfernt. Die Blitzsäulenchromatographie (Elutionsmittel : Lösungsmittelbenzin, Siedepunkt: 40–60°C : Ethylacetat 95 : 5) ergab 2C4 (83 mg, 83%) und 2C5 (10, 0 mg, 10%).
- NMR-Daten für 2C4
- 1H NMR (CDCl3, 300 MHz) δH 3.30 (2H, br s, HC + CCH 2), 3.69 (2H, ab q, J = 7.43 Hz COCH 2), 5.38 (1H, d, J = 25.23 Hz CHCOCH2), 6.94 (1H, d, J = 7.47 Hz 1 × Ar-H), 7.30 (8H, br m, 7 × Ar-H & 1 × C=CHCH2).
13C NMR (CDCl3, 75.47 MHz) δC 37.6 & 43.5 (2 × CH2), 54.3 (CHC=CHCH2), 120.6, 123.7, 124.7, 125.0, 125.1, 126.5, 127.5, 127.6, 136.5 (8 × Ar-CH & 1 × C=CH), 137.4, 141.7, 143.0, 143.4, 14.0, (4 × Ar-c & × C=CH) 215.7 (C=O). - Synthese von 2C6 2C6 : 1-(2-Indenyl)-1-prop-2-enyl-2-indanon

- Bei der Synthese wird die gleiche Prozedur wie bei 2C3 angewendet, allerdings wird Allylbromid anstatt Ethyljodid verwendet.
1H NMR (CDCl3, 300 MHz) δH.
13C NMR (CDCl3, 75.47 MHz) δC 38.3, 41.1, 41.9, 44.2 (4 × CH 2), 57.3, 50.2 (2 × CH), 118.6, 124.5, 126.2, 127.7, 130.5, 132.8, 113.6, 133.7 (8 × Ar-H) 143.0, 143.7, 143.9. 148.6 (4 × Ar-C), 217.8 (C=O). Synthese von 2C7 Alkylierung von 2C1Ausbeute (57 mg, 42%)
1H NMR (CDCl3, 300 MHz) δH 3.22–3.80 (6H, m, CH 2's), 6.63 (1H, s, CH=C), 6.83–7.42(8H, m, 8 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 38.9, 42.8, 43.6 (3 × CH2), 63.4 (qC), 120.8, 123.6, 124.6, 124.7, 125.8, 126.3, 126.3, 126.4, 126.4, 127.4, 127.4, 127.7, 127.8, 129.5, 130.2 (13 × Ar-CH & 2 × C=CH), 136.1, 136.7, 143.3, 143.5, 149.2 (5 × Ar-C), 216.3 (C=O). - Synthese von 2C8
- 2C7 (100 mg, 0,3 mMol) wurde in Ethylacetat : Ethanol (2 : 1, 10 ml) gelöst, und in diese Lösung wurde NaBH4 (100 mg) gegeben. Man ließ die Reaktion bei Raumtemperatur für 2 Stunden stehen. Das Produkt wurde in Ethylacetat extrahiert, und das Produkt wurde über die Blitzsäulenchromatographie gereinigt und als Mischung aus den Diastereomeren 2C8 (0,067 g, 66,1 ) isoliert.
-

- Synthese von 2C10

- 2C8 (100 mg, 0,3 mMol) wurden in sauberem, trockenen DCM (5 ml) dispergiert. Dazu wurden Triethylamin (0,1 ml), Essigsäureanhydrid (0,25 ml) und DMAP (90,05 g) gegeben. Man ließ die Reaktion bei Raumtemperatur für 15 Minuten rühren. Das Rohraktionsgemisch wurde dann durch eine Säule mit Petroleumether : Ethylacetat, 9 : 1, gegeben.
1H NMR (CDCl3, 300 MHz) δH 2.34 (3H, 2 × s, COCH), 2.89–3.65 (6H, m, 3 × CH 2), 5.75 (1H, m, CHOAc), 6.93 (1H, t, J = 2.2 Hz, C=CH), 6.95–7.52 (13H, m, Ar-CH).
13C NMR (CDCl3, 75.47 MHz) δC 20.6, 21.0, (COCH3), 36.8, 37.4, 39.1, 39.4, 40.4, 40.6 (3 × CH2) 57.2, 58.7 (qC), 79.40, 80.9 (CHOH), 120.4, 120.5, 123.3, 124.1, 124.2, 124.5, 124.7, 125.5, 125.9, 126.0, 126.1, 126.2, 126.4, 126.6, 127.3, 127.3, 127.4, 127.5, 127.8, 127.7, 128.4, 128.4, 129.5, 129.8, 130.3, 130.8, (13 × Ar-CH), 136.8, 137.5, 139.5, 140.2, 140.7, 142.7, 142.8, 143.3, 144.3, 144.9, 145.1, 150.6, 151.7, (5 × Ar-C & 1 × C=CH) 170.1, 170.2, (COCH3). - Verknüpfungsreaktion des entsprechenden Silylenolethers von Indan-1-on an das entsprechende Dimethylacetal von Indan-2-on Synthese eines Silylenolethers von Indan-1-on

- In eine gerührte Lösung aus Indan-1-on (1,0 g, 7,57 mMol) und Triethylamin (0,84 g, 1,16 ml, 8,32 mMol) in Dichlormethan bei 0°C wurde Trimethylsilyltrifluormethansulfonat (1,68 g, 1,36 ml, 7,58 mMol) gegeben. Man ließ die Lösung bei 0°C für 14 Minuten rühren, und dann wurde die Lösung schnell durch eine Silikasäule mit Petroleumether (Siedepunkt 40–60°C) : Ethyl als Elutionsmittel gegeben. Der Silylenolether wurde als klares farbloses Öl (5) (1,0 g, 77,0%) isoliert.
- Synthese des Dimethylacetals von Indan-2-on

- In eine gerührte Lösung aus Indan-2-on (1,0 g, 7,57 mMol) in Methanol (12 ml) wurden Trimethylorthoformat (2 ml) und p-Toluolsulfonsäure (etwa 1 Mol-%) gegeben. Man ließ dann die Lösung bei Raumtemperatur für 2 Stunden rühren. In diese Lösung wurde dann festes Natriumbicarbonat (etwa 0,50 g) gegeben. Das Methanol wurde aus dem Reaktionsgemisch verdampft. Der Rohfeststoff war dann zwischen Ether : Wasser (1 : 1) (50 ml) verteilt. Die organische Schicht wurde isoliert und die wässrige Schicht mit Ether (3 × 20 ml) extrahiert. Die vereinten organischen Schichten wurden mit Natriumsulfat getrocknet. Nach der Verdampfung des Lösungsmittels ließ man dann das Rohprodukt durch eine Silikasäule mit Petroleumether zu 100% in der Abstufung Petroleumether : Ethylacetat, 100 : 1 gehen. Nach der Verdampfung des Elutionsmittels wurde das Dimethylacetal von Indan-2-on als klares farbloses Öl (0,80 g, 60%) isoliert.
1H NMR (CDCl3, 300 MHz) δH 3.21 (4H, s, 2 × CH 2), 3.35 (6H, s, 2 × ? OCH 3), 7.22 (4H, s, 4 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 41.2, 41.2 (CH2), 49.4, 49.4 (2 × OCH3), 111.4 (C(Ome)2), 124.5, 126.5 (4 × Ar-CH), 139.8 (2 × qC). - Synthese von 3C1

- In eine gerührte Lösung aus dem Silylenolether von Indan-1-on (5) (0,80 g, 3,92 mMol) und dem entsprechenden Dimethylacetal von Indan-2-on (0,80 g, 3,92 mMol) in Dichlormethan bei –78°C wurde eine katalytische Menge an TMS-Triflate gegeben. Die Lösung ließ man bei –78°C für 3 Stunden rühren, und dann ließ man sie innerhalb einer Stunde –50°C erreichen. In diese Lösung wurde dann eine 5%ige Lösung auf Natriumdicarbonat (etwa 20 ml) gegeben. Die organische Schicht wurde isoliert und die wässrige Schicht mit Dichlormethan (2 × 20 ml) extrahiert. Die vereinten organischen Schichten wurden mit Natriumsulfat getrocknet. Nach der Verdampfung des Lösungsmittels ließ man das Rohprodukt durch eine Silikasäule mit Petroleumether zu 100 in der Abstufung Petroleumether : Ethylacetat, 100 : 4 als Elutionsmittel gehen. Nach der Verdampfung des Elutionsmittels wurde das 3C1 als leicht gefärbtes Öl (0,50 g, 50,5%) isoliert.
- Bei der Zugabe von Ether zu dem Öl kristallisierte das 3C1 als weiße Kristalle aus.
1H NMR (CDCl3, 300 MHz) δH 3.06 (3H, s, OCH), 3.10 (1H, m, CH), 3.37 (2H, q, J = 17, CHCH 2), 3.30 (2H, br t, CH 2), 7.16 (4H, m, 4 × Ar-H), 7.57 (1H, t, 1 × Ar-H), 7.59 (1H, t, 1 × Ar-H), 7.73 (1H, d, 1 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 29.7, 40.6, 41.7 (3 × CH2), 51.1 (CH), 53.5 (OCH3), 87.4 (qC), 123.9, 124.1, 124.3, 126.4, 126.49, 126.52, 127.2, 134.7 (8 × Ar-CH), 137.7, 140.9, 141.5, 153.7 (4 × Ar-C), 206.3 (C=O). - Synthese von 3C2

- 3C1 (200 g, 0,689 mMol) wurde in Methanol (3 ml) und DCM (1 ml) gelöst, in diese Lösung wurde unter Rühren Trifluormethansulfonsäure (Triflic Acid) (45 μl) gegeben. Das Reaktionsgemisch ließ man für 1 Stunde unter Rückfluss, wobei sich ein Niederschlag bildete. Die Lösung wurde dann in einem Eisbad abgekühlt, filtriert und der Feststoff wurde getrocknet. Die Analyse dieses gelben Feststoffes ergab das 3C2 (100 mg, 50 %).
1H NMR (CDCl3, 300 MHz) δH 3.65, 3.90, 4.40 (6H, 3 × br s, 3 × CH 2), 7.20 (2H, m, 2 × Ar-H), 7.35 (3H, m, 3 × Ar-H), 7.52 (2H, m, 2 × Ar-H), 7.84 (1H, d, J = 7.7 Hz, 1 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 31.9, 39.3, 40.2 (3 × CH2), 123.8, 124.4, 124.9, 126.1, 126.6, 126.9, 127.3, 135.4 (8 × Ar-CH) 129.1, 139.1, 139.7, 142.2, 148.4, 154.8 (4 × Ar-C & 2 × C=C), 193.5 (C=O). - Herstellung von 3C3

- 1H NMR (CDCl3, 300 MHz) δH 2.62–2.90 (2H, m, CH2=CHCH 2), 3.28–3.68 (4H, m, 2 × CH2), 4.99–5.21 (2H, m, CH-CH2), 6.69 (1H, s, C-CHCH2), 7.05–7.30 (3H, m, 3 × Ar-H), 7.34–7.42 (2H, m, 2 × Ar-H), 7.48–7.56 (1H, dd, J = 87 Hz, 7.3 Hz 1 × Ar-H), 7.56–7.67 (1H, m, 1 × Ar-H), 7.71–7.79 (1H, m, 1 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 38.3, 38.6, 41.3 (3 × CH2), 55.8 (qC), 118.7 (CH-CH2), 120.5, 123.4, 124.7, 126.3, 127.6, 127.9, 133.7, 135.1, 135.3 (8 × Ar-CH & 1 × CH = CH2), 143.2, 144.2, 148.9, 152.5 (4 × Ar-C), 205.9 (C=O). - Synthese von 3C4 Reduktion von 3C3

- 3C3 (100 mg, 0,351 mMol) wurde in Ethanol (20 ml) und Ethylacetat (10 ml) gelöst. In diese Lösung wurde unter Rühren der Wilkinson-Katalysator (0,1 g) gegeben. Die Reaktionsmischung dann unter Wasserstoff für 20 Stunden gerührt. Eine weitere Menge des Wilkinson-Katalysators (200 mg) wurde dann hinzugegeben. Man ließ die Reaktion unter Wasserstoff für weitere 12 Stunden rühren. Das Lösungsmittel wurde dann entfernt, und das Rohprodukt wurde über die Blitzsäulenchromatographie gereinigt, wobei das 3C4 (90 mg, 90%) erhalten wurde.
1H NMR (CDCl3, 300 MHz) δH 1.00 (3H, t, J = 1.6 Hz, CH2CH2CH 3), 1.33 (2H, m, CH2CH 2CH3), 1.90 (1H, m, CH of CH 2CH2CH3), 2.10 (1H, m, Ch of CH 2CH2CH3), 3.45 (2H, ab q, CH=C-CH 2), 3.54 (2H, ab q, J = 17.7 Hz, C-CH 2), 6.71 (1H, s, CH=C-CH2), 7.14 (1H, dt, J = 1.5 Hz & 5.7 Hz, Ar-H), 7.25 (2H, br m, 2 × Ar-H), 7.36 (2H, br t, 2 × Ar-H), 7.52 (1H, br d, Ar-H, 7.63 (1H, dt, J = 1.0 Hz, ar-H), 7.77 (1H, br d, J = 7.7 Hz, Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 14.5 (CH3), 18.3, 38.6, 39.0, 39.5 (4 × CH2), 56.5 (qC), 120.5, 123.5, 124.7, 125.1, 126.2, 126.2, 127.0, 127.7, 134.9 (8 × Ar-CH & 1 × C=CH), 135.5, 143.3, 144.4, 149.5, 152.5 (4 × Ar-C & 1 × CH=C), 206.6 (C=O). - Synthese von 3C5 Alkylierung von 3C1

- 3C1 (400 mg, 1,44 mMol) wurde in Ether (12 ml) und tButanol (2 ml) gelöst, dazu wurde dann Benzylbromid (1,0 g, 0,66 ml, 5,76 mMol) gegeben. In diese Lösung wurde Kalium-tert.-butoxid (160 mg, 1,44 mMol) in tButanol (7 ml) tropfenweise unter Rühren über 20 Minuten gegeben. Man ließ die Lösung für 3 Stunden rühren. In diese Lösung wurde eine gesättigte wässrige Ammoniumchloridlösung (20 ml) gegeben, und die organische Phase wurde mit Ether (2 × 50 ml) extrahiert. Die organischen Schichten wurden vereint, getrocknet, und das Rohprodukt wurde über die Blitzsäulenchromatographie gereinigt, und man erhielt das 3C5 (388 mg, 80%).
1H NMR (CDCl3, 300 MHz) δH 3.48 (2H, ab q, J = 13.0 Hz, CH 2), 3.45 (2H, d, J = 7.4 Hz, CH 2), 3.65 (2H, d, CH 2), 6.78 (1H, d, J = 0.7 Hz, CH=C), 7.22–7.45 (11H, br m, 11 × Ar-H), 7.54 (1H, dt, J = 1.2 Hz & 7.6 Hz, Ar-H, 7.81 (1H, d, J = 7.2 Hx, Ar=H).
13C NMR (CDCl3, 75.47 MHz) δC 37.5, 38.5, 38.8, 42.2 (3 × CH2), 57.2 (qC), 120.5, 123.4, 124.1, 124.3, 124.5, 125.9, 126.2, 126.4, 128.1, 129.9, 134.8, (13 × Ar-CH & 1 × C=CH), 135.1, 137.3, 143.1, 144.1, 149.1, 152.4 (5 × Ar-C & 1 × C=CH), 205.9 (C=O). - Synthese von 3C6 & 3C7 Natriumborhydridreduktion von 3C5

- 3C5 (0,50 g, 1,48 mMol) wurde in Ethylacetat : Ethanol (2 : 1, 21 ml) gelöst und es wurde Natriumborhydrid (0,50 g, 13,15 mMol) in die Reaktion gegeben. Man ließ die Reaktion bei Raumtemperatur für 3 Stunden rühren. Nach der Verdampfung des Lösungsmittels blieb ein weißer Feststoff, zu dem DCM (2 ml) gegeben wurde, und man ließ die Aufschlämmung durch eine Silikasäule mit Lösungsmittelbenzin (Siedepunkt 40–60°C) : Ethylacetat, 100 : 1 als Lösungsmittel gehen. Das erste Paar der Enantiomeren 3C6 wurde eluiert, und die Verdampfung des Lösungsmittels ergab einen weißen Feststoff (0,24 g, 96%). Das zweite Enantionerenpaar 3C7 wurde dann eluiert, und die Verdampfung des Lösungsmittels ergab ein mobiles Öl (0,24 g, 96%).
- 3C6
- 1H NMR (CDCl3, 300 MHz) δH 2.99 2H, dd, CH 2), 3.21 (2H, dd, CH 2), 3.45 (2H, ab q, CH 2), 5.05 (1H, m, CHOH), 6.68 (1H, 2, J = 0.5 Hz, CH=C-CH2), 6.96 (2H, m, 2 × Ar-H), 7.16–7.45 (11H, br m, 11 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 38.4, 40.4, 43.4 (3 × CH2), 56.4 (1 × qC), 81.8 (1 × CHOH), 120.6, 123.5, 124.3, 124.9, 125.0, 126.3, 126.3, 126.9, 128.0, 128.0, 128.7, 130.2, 130.2, 130.4 (13 × Ar-CH & 1 × C=CH), 138.1, 141.7, 143.3, 143.7, 144.5, 151.0 (5 × Ar-C & 1 × C=CH). - 3C7
- 1H NMR (CDCl3, 300 MHz) δH 2.32 (1H, br m, CHOH), 2.75 & 3.20 (2H, dd, J = 13.4 Hz, CH 2), 3.17 (2H, ab q, J = 15.7 Hz, CH 2), 3.55 (2H, ab q, J = 22.6 Hz, CH 2), 5.25 (1H, br s, CHOH), 6.52 (1H, d, J = 0.4 Hz, C=CH), 6.91 (2H, dd, 2 × Ar-H), 7.20 (4H, br m, 4 × Ar-H), 7.30 (5H, br m, 5 × Ar-H), 7.50 (2H, br m, 2 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 29.8, 38.8, 40.4 (3 × CH2), 55.8 (qC), 83.8 (CHOH), 120.4, 123.5, 123.9, 124.0, 124.9, 126.0, 126.3, 126.8, 127.7, 127.9, 128.3, 128.6, 130.1, 130.4 (13 × Ar-CH & 1 × C=CH), 138.3, 140.6, 142.9, 143.8, 144.7 153.2 (5 × Ar-CH 1 × C=CH). - Synthese von 3C8 und 3C9 durch Reduktion mit Lithium-tri-tert.-butoxyaluminohydrid

- 3C5 (200 mg, 0,593 mMol) wurde in trockenem THF (5 ml) gelöst, und dann wurde Lithium-tri-tert.-butoxyaluminohydrid (0,50 g, 1,97 mMol) hinzugefügt. Man ließ die Lösung für 3 Stunden rühren. Das Lösungsmittel wurde entfernt, und das Rohreaktionsgemisch wurde filtriert und über die Blitzsäulenchromatographie gereinigt, wobei man das 3C8 (90 mg, 96%) und das 3C9 (90 mg, 96%) erhielt.
- 3C8
- 1H NMR (CDCl3, 300 MHz) δH 2.99 (2H, dd, J = 13.7 Hz, CH 2), 3.21 (2H, t, J = 4.4 Hz, CH 2), 3.45 (2H, abq, J = 22.6 Hz, CH 2), 5.05 (1H, m, CHOH), 6.68 (1H, s, CH=C-CH2), 6.96 (2H, m, 2 × Ar-H), 7.16–7.45 (11H, br m, 11 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 38.4, 40.4, 43.4 (3 × CH2), 56.4 (qC), 81.8 (1 × CHOH), 120.6, 123.5, 124.3, 124.9, 125.0, 126.3, 126.3, 126.9, 128.0, 128.0, 128.7, 130.2, 130.2, 130.4, (13 × Ar-CH & 1 × C=CH), 138.1, 141.7, 143.3, 143.7, 144.5, 151.0 (5 × Ar-C & 1 × C=CH). - 3C9
- 1H NMR (CDCl3, 300 MHz) δH 2.32 (1H, br m, CHOH), 2.99 (2H, dd, J = 13.4 Hz, CH=C-CH 2), 3.17 (2H, ab q, J = 15.7 Hz, CH), 3.55 (2H, ab q, J = 22.6 Hz, CH 2), 5.25 (1H, br s, CHOH), 6.52 (1H, d, J = 0.4 Hz, C=CH), 6.91 (2H, dd, 2 × Ar-H), 7.20 (4H, br m, 4 × Ar-H), 7.30 (5H, br m, 5 × Ar-H), 7.50 (2H, br m, 2 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 38.4, 38.5, 40.0 (3 × CH2), 55.8 (qC) 83.8 (CHOH), 120.4, 123.5, 123.9, 124.0, 124.9, 126.0, 126.3, 126.8, 127.7, 127.9, 128.3, 128.6, 130.1, 130.4 (13 × Ar-CH), 138.3, 140.6, 142.9, 143.8, 144.7 153.2 (5 × Ar-C-CH & 1 × C=CH), 138.3, 140.6, 142.9, 143.8, 144.7, 153.2 (5 × Ar-C & 1 × C=CH). - Synthese von 3C10

- 3C8 & 3C9 (50 mg, 0,15 mMol) wurden in sauberem, trockenen DCM (5 ml) gelöst. In diese Lösung wurden Triethylamin (0,1 ml), DMAP (0,05 g) und Essigsäureanhydrid (0,25 ml, 10 Äquivalente) gegeben. Man ließ das Reaktionsgemisch bei Raumtemperatur für 15 Minuten rühren und durch eine Säule bei Raumtemperatur für 15 Minuten gehen und durch eine Silikasäule mit Lösungsmittelbenzin (Siedepunkt 40–60°C) : Ethylacetat (8 : 2) als Elutionsmittel gehen, und man erhielt das 3C10 (45 mg, 80,05%).
1H NMR (CDCl3, 300 MHz) δH 2.24 (3H, s, COCH 3), 3.05–3.40 (6H, m, 3 × CH 2), 6.40, 6.53 (2H, 2 × s, C=CH und CHOCOCH3), 6.93–6.95 (2H, m, 2 × Ar-H), 7.13–7.17 (11H, m, 11 × Ar-H)
13C NMR (CDCl3, 300 MHz) δC 21.3 (OCOCH3), 39.6, 40.6, 40.7 (3 × CH2), 54.4 (qC), 82.8 (CHOCOCH3), 120.5, 123.4, 124.2, 124.5, 125.7, 126.2, 126.3, 126.8, 127.9, 127.9, 128.9, 128.9, 129.4, 129.9, (13 × Ar-CH, Vinyl-CH), 138.1, 140.6, 142.5, 142. 7, 144.5, 151.8, (5 × Ar-C und 1 × CH=CH2), 170.8 (OCOCH3) - Synthese von 3C11 Alkylierung von 3C1

- 3C1 (200 mg, 0, 719 mMol) wurde in Ether (6 ml) und tButanol (1 ml) gelöst, in diese Lösung wurde Methyl-4-(brommethyl)benzoat (660 mg, 2,88 mMol) gegeben. Kalium-tert.-butoxid (80 mg, 0,719 mMol) wurde in tBuOH (6 ml) und Ether (1 ml) gelöst. Die tBuOK-Lösung wurde über einen Zeitraum von 3 Stunden hinzugegeben. Man ließ die Lösung für weitere 2 Stunden rühren. In diese Lösung wurde wässriges Ammoniumchlorid (20 ml) gegeben. Die organische Phase wurde mit Ether extrahiert, und das Rohreaktionsgemisch wurde über die Blitzsäulenchromatographie gereinigt, wobei man das 3C11 (230 mg, 82%) erhielt.
1H NMR (CDCl3, 300 MHz) δH 3.29–3.61 (6H, m, 3 × CH 2), 3.84 (3H, s, CO2CH 3), 6.73 (1H, br s, CH=CCH2), 7.14 (1H, dt, J = 1.5 Hz & 7.2 Hz, Ar-H), 7.20 (6H, m, 6 × Ar-H), 7.35 (1H, d, J = 7.6 Hz, 1 × Ar-H), 7.53 (1H, dt, J = 1.2 Hz & 7.4 Hz, Ar-H), 7.75 (1H, d, J = 7.6 Hz, Ar-H, 7.85 (2H, d, × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 37.5, 38.6, 42.1 (3 × CH2), 51.8 (CO2 CH3), 56.9 (qC), 120.6, 123.4, 124.5, 124.6, 126.0, 126.3, 127.6, 128.3, 128.5, 129.3, 129.3, 129.9, 129.9, 135.0, (12 × Ar-CH & 1 × Ar-C & 1 × C=CH) 142.8, 142.9, 143.9, 148.6, 142.1 (5 × Ar-C), 166.7 (C)O2CH3), 205.5 C=O). - Synthese von 3C12 Kupplungsreaktion Silyenolether von 4-Methoxy-1-indanon

- In eine gerührte Lösung aus 4-Methoxy-1-indanon (200 mg, 1,24 mMol) und Triethylamin (0,15 g, 0,21 ml, 1,48 mMol) in Dichlormethan bei 0°C wurde Trimethylsilyltrifluormethansulfonat (0,27 g, 0,22 ml, 1,24 mMol) gegeben. Man ließ die Lösung bei 0°C für 14 Minuten rühren, und dann wurde die Lösung schnell durch eine Silikasäule mit Petroleumether (Siedepunkt 40– 60°C) : Ethylacetat 100 : 0,5 als Elutionsmittel gelassen. Nach der Verdampfung des Elutionsmittels wurde der Silylenolether (1) als klares farbloses Öl (260 mg, 91%) isoliert.
- Synthese von 3C12

- In eine gerührte Lösung aus dem Silylenolether von 4-Methoxy-1-indanon (200 mg, 0,854 mMol) und dem entsprechenden Dimethylacetal von Indan-2-on (180 mg, 1,03 mMol) in Dichlormethan bei –78°C wurde eine katalytische Menge von TMS-Triflate (45 μl) gegeben. Man ließ die Lösung bei –78°C für 3 Stunden rühren und dann innerhalb einer Stunde –50°C erreichen. In diese Lösung wurde dann festes Natriumbicarbonat (etwa 2,0 g) gegeben. Die organische Schicht wurde dekandiert und der Feststoffrückstand mit Dichlormethan (2 × 20 ml) extrahiert. Die vereinten organischen Schichten wurden mit Natriumsulfat getrocknet. Nach der Verdampfung des Lösungsmittels ließ man das Rohprodukt durch eine Silikasäule mit Petroleumether zu 100 in der Abstufung Petroleumether : Ethylacetat, 100 : 4, als Elutionsmittel laufen. Nach der Verdampfung des Elutionsmittels wurde das 3C12 als leicht gefärbtes Öl (0,16 g, 61%) isoliert. Bei der Zugabe von Ether zu dem Öl kristallisierte das 3C12 als weiße Kristalle auf.
- 3Cl2
- 1H NMR (CDCl3, 300 MHz) δH 3.06 (3H, s, CH2C-OCH 3), 3.07 (2H, m, CH 2), 3.25 (4H, m, 1 × CH of CH 2 & 1 × CHCH2 1 × CH 2), 3.50 (1H, d, J = 17.0 Hz, CH of CH 2), 3.92 (3H, s, ArOCH 3), 7.03 (1H, t, J = 4 Hz, Ar-H), 7.15 (4H, m, 4 × Ar-H), 7.38 (2H, d, J = 5.0 Hz, 2 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 26.5, 40.5, 41.6 (3 × CH2), 51.0, 53.4, 55.4 (2 × OCH3 & 1 × CH), 87.4 (qC), 114.5, 115.2, 115.3, 124.1, 124.2, 126.4, 128.7 (7 × ar-CH), 139.1, 140.9, 141.5, 142.5, 156.8 (5 × Ar-C), 206.3 (C=O). - Synthese von 3C13 Alkylierung von 3C12

- Übliche Prozedur für die Alkylierung von β-Methoxycarbonylverbindungen. Das 3C13 wurde isoliert (160 mg, 80%).
1H NMR (CDCl3, 300 MHz) δH 3.26 (2H, ab q, J = 17.8 Hz, CH 2), 3.45 (4H, m, 2 × CH 2), 3.85, 3.86 (6H, 2 × s, CO2CH 3 & Ar-OCH 2), 6.73 (1H, br s, C=CH), 6.98 (1H, m, Ar-H), 7.21 (1H, 7.83 (2H, br d, 2 × Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 34.2, 38.8, 42.2 (3 × CH2), 51.9, 55.4 (1 × OCH3 & CO2 CH3), 56.9 (qC), 115.2, 116.1, 120.7, 123.5, 124.5, 126.3, 128.5, 129.2, 129.4, 129.4, 130.1, 130.1 (11 × Ar-CH & 1 × CH=C), 128.4, 136.6, 140.9, 142.9, 143.1, 144.1, 148.7, 156.4 (7 × Ar-C & 1 × C=Ch), 166.9 (CO2Ch3), 205.8 (C=O). - Synthese von 3C17 Synthese des Silylenolethers von Bromindanon

- Zu einer gerührten Lösung aus 6-Brom-5,7-dimethyl-1-indanon (0,5 g, 2,11 mMol) in DCM (3 ml) bei 0°C wurde Triethylamin (0,21 g, 0,29 ml, 2,08 mMol) und TMS-Triflate (0,466 g, 0,38 ml, 2,09 mMol) gegeben. Man ließ die Lösung bei 0°C für 10 Minuten rühren und dann die Lösung durch eine Silikasäule mit Petroleumether als Elutionsmittel laufen, und man erhielt den Silylenolether von 6-Brom-5,7-dimethyl-1-indanon als mobiles Öl (0, 62 g, 95%).
- Synthese des Brommethoxydimeren

- Zu einer Lösung aus dem Dimethylacetal von 2-Indanon (0,50 g, 2,81 mMol) und dem Silylenolether von 6-Brom-5,7-dimethyl-1-indanon (0,50 g, 1,76 mMol) in DCM bei –78°C wurde eine katalytische Menge an TMS-Triflate (40 μl) gegeben. Die Reaktion ließ man dann langsam auf Raumtemperatur erwärmen und dann bei Raumtemperatur über 4 Stunden rühren. In diese Lösung wurde bei Raumtemperatur festes Natriumbicarbonat (1,0 g) gegeben, und man ließ die heterogene Mischung für 15 Minuten rühren. Die Mischung wurde dann durch eine Silikasäule gelassen, und das Brommethoxydimer 3C17 wurde als gelblicher Feststoff isoliert, der schnell aus Diethylether unter Bildung weißer Kristalle der Titelverbindung (0,3 g, 49,2%) auskristallisierte.
1H NMR (CDCl3, 300 MHz) δH 2.50 (3H, s, Ar-CH3), 2.80 (3H, s, Ar-CH 3), 2.95 (3H, s, OCH 3), 3.18 (1H, br m, COCHCH2), 3.16 (2H, br m, CH 2), 3.28 (2H, br m, CH 2), 3.30 (2H, ab q, J = 12.6 Hz, CCH 2), 7.15 (4H, m, 4 × Ar-H), 7.24 (1H, br s, Ar-H).
13C NMR (CDCl3, 75.47 MHz) δC 17.5, 25.1 (2 × CH3), 29.6, 40.6, 41.6 (3 × CH2), 51.1, 54.2 (1 × CH & 1 × OCH3), 87.4 (qC), 124.1, 124.3, 125.7, 126.5, 128.1, 134.1, 139.0, 140.9, 141.5, 141.8, 144.8, 153.1 (5 × Ar-CH, 4 × Ar-C & 2 × ArC-CH3, 1 × ArCBr), 205.7 (C=O). - Synthese von 3C18 Alkylierung von 3C17

- Zu einer Lösung von Brommethoxy 3C17 (200 mg, 0,519 mMol) in Ether (6 ml) wurde Benzylbromid (0,30 g, 0,20 ml, 1,75 mMol) gegeben. In diese Lösung wurde bei Raumtemperatur tropfenweise eine Lösung aus Kalium-tert.-butoxid (0,05 g, 0,439 mMol) in tBuOH (6 ml) gegeben. Die Analyse dieser Lösung zeigte, dass das Brommethoxy 3C17 insgesamt in das Brombenzyl 3C18 mit einem zweiten Produkt mit einem etwas niedrigeren Rf-Wert als das Brommethoxy 3C18 als Ausgangsmaterial umgewandelt war. Das Rohreaktionsgemisch wurde über die Blitzsäulenchromatographie gereinigt und man erhielt das 3C18 (0,12 g, 52%).
- Synthese von 3C19 & 3C20 durch die Reduktion mit Lithium-tri-tert.-butoxyaluminohydrid

- 3C18 (200 mg, 0,451 mMol) wurde in trockenem THF (5 ml) gelöst, und dazu wurde Lithium-tri-tert.-butoxyaluminohydrid (0,50 g, 1,97 mMol) gegeben. Man ließ die Lösung für 3 Stunden rühren. Das Lösungsmittel wurde entfernt, und das Rohreaktionsgemisch wurde über die Blitzsäulenchromatographie gereinigt, und man erhielt die beiden Diastereomeren 3C19 (90 mg, 96%) und 3C20 (90 mg, 96%).
- 3C20
- 1H NMR (CDCl3, 300 MHz) dH 2.40 (3H, s, CH 3), 2.48 (3H, s, CH 3), 2.80 (1H, d, J = 13.6 Hz, CH of CH2), 3.00 (2H, ab q, J = 15.6 Hz, CH of CH2), 3.23 (1H, d, J = 13.6 Hz, CH of CH2), 3.54 (2H, ab q, J = 22.6 Hz, CH2), 5.18 (1H, br m, CHOH), 6.52 (1H, br s, C=CH), 6.89 (2H, m, 2 × Ar-H), 7.20 (5H, m, 5 × Ar-H), 7.30 (3H, 2 × Ar-H), 7.45 (1H, d, J = 6.0 Hz, 1 × Ar-H).
- Synthese von 3C21 Verknüpfungsreaktion

- Zu einer gerührten Lösung aus 5-Brom-4,6-dimethyl-1-indanon (0,5 g, 2,11 mMol) in DCM (3 ml) bei 0°C wurden Triethylamin (0,21 g, 0,29 ml, 2,08 mMol) und TMS-Triflate (0,466 g, 0,38 ml, 2,09 mMol) gegeben. Man ließ die Lösung bei 0°C für 10 Minuten rühren und dann wurde die Lösung durch eine Silikasäule mit Petroleumether als Elutionsmittel gegeben, und man erhielt den Silylenolether von 5-Brom-4,6-dimethyl-1-indanon als weißen kristallinen Feststoff (0,60 g, 94%).
- Synthese des Brommethoxydimeren 3C21

- Zu einer Lösung aus dem Dimethylacetal des 2-Indanons (0,50 g, 2,81 mMol) und dem Silylenolether von 5-Brom-4,6-dimethyl-1-indanon (0,50 g, 1,76 mMol) in DCM bei –78°C wurde eine katalytische Menge TMS-Triflate (40 μl) gegeben. Die Reaktion ließ man dann langsam auf Raumtemperatur erwärmen und dann bei Raumtemperatur für 4 Stunden rühren. In diese Lösung wurde festes Natriumbicarbonat (1,0 g) gegeben, und man ließ die heterogene Mischung für 15 Stunden rühren. Die Mischung wurde dann durch eine Silikasäule gelassen, um das Brommethoxy 3C21 als gelblichen Feststoff zu isolieren, der schnell aus Diethylether unter Erhalt feiner weißer Kristalle (271 mg, 40%) auskristallisierte.
1H NMR (CDCl3, 300 MHz) δH 2.54 (6H, br s, 3 × überlapp. CH 3), 3.10 (3H, s, OCH 3), 3.12 (1H, m, CHCH2), 3.21–3.38 (5H, m, 2 × CH 2 & 1 × CH of CH 2), 3.73 (1H, d, CH of CH 2), 7.16 (4H, m, 4 × Ar-H), 7.44 (1H, s, Ar-H). - Synthese von 3C22

- Zu einer Lösung des Brommethoxydimers 3C21 (200 mg, 0,519 mMol) in Ether (6 ml) wurde Benzylbromid (0,30 g, 0,20 ml, 1,75 mMol) gegeben. In diese Lösung bei Raumtemperatur wurde tropfenweise eine Lösung aus Kalium-tert.-butoxid (0,05 g, 0,439 mMol) in tBuOH (6 ml) gegeben. Die Analyse dieser Lösung zeigte, dass das Brommethoxydimer insgesamt in das Brombenzyldimer 3C22 und ein Produkt mit einem etwas niedrigeren Rf als das Ausgangsbrommethoxydimer umgewandelt war. Das Bromreaktionsgemisch wurde über die Blitzsäulenchromatographie gereinigt, und man erhielt das 3C22 (0,14 g, 61%).
- Synthese von 3C23 & 3024 durch Reduktion mit Lithium-tri-tert.-butoxyaluminohydrid

- 3C22 (200 mg, 0,451 mMol) wurde in trockenem THF (5 ml) gelöst, und dazu wurde Lithium-tri-tert.-butoxyaluminohydrid (0,50 g, 1,97 mMol) gegeben. Man ließ die Lösung für 3 Stunden rühren. Das Lösungsmittel wurde entfernt, und das Bromreaktionsgemisch wurde über die Blitzsäulenchromatographie gereinigt und man erhielt das 3C23 (90 mg, 96%) und das 3C24 (90 mg, 96 %).
- Synthese von 3C31
- Gleiche Prozedur wie für die Synthese 3C5.
- Synthese von 3C22 & 3C33

- Zu einer Lösung des Benzyldimers 3031 (0,10 g, 0,27 mMol) in THF (4 ml) wurde unter Rühren bei 0°C Lithium-tri-tert.-butoxyaluminohydrid (0,20 g, 0,79 mMol) gegeben. Man ließ die Mischung bei 0°C für 1 Stunde rühren und dann bei Raumtemperatur für 2 Stunden. In diese Lösung wurde dann (80 mg, 0,31 mMol) Lithium-tri-tert.-butoxyaluminohydrid gegeben, und man ließ die Mischung bei Raumtemperatur für 3 Tage rühren. Das Lösungsmittel wurde dann verdampft und der Rückstand wurde in DCM (2 ml) aufgenommen. Die trübe Mischung wurde dann durch eine Silikasäule laufen gelassen, um das Lithium-tri-tert.-butoxyaluminohydrid zu entfernen. Das Elutionsmittel, das das Gemisch aus den Alkoholen enthielt, wurde bis zur Trockne verdampft, wobei ein Öl hinterlassen wurde, das dann in einer minimalen Menge DCM aufgenommen wurde. Die beiden Paare aus den diastereomeren Alkoholen wurden voneinander über die Blitzsäulenchromatographie getrennt, wobei beide Alkoholpaare als mobile Öle 3C32 und 3C33 mit einer Gesamtausbeute (35 mg, 95%) erhalten wurden.
- Top Spot
- 1H NMR (CDCl3, 300 MHz) δH 2.38, 2.45 (6H, 2 × s, 2 × CH 3), 2.80 (2H, ab q, J = 13.4 Hz, CH 2), 3.00 (2H, ab q, J = 15.9 Hz, CH 2), 3.65 (2H, ab q, CH 2), 5.03 (1H, s, CHOH), 6.58 (1H, s, C=CH), 6.80 (2H, br m, 2 × Ar-H), 6.93 (1H, s, 1 × Ar-H), 7.15– 7.23 (6H, br m, 6 × Ar-H), 7.47 (1H, d J = 3.0 Hz, 1 × Ar-H).
- Es sollte selbstverständlich sein, dass die Verbindungen die pharmakologisch annehmbaren Salze, Ester, Isomere und Solvate davon umfassen. Ein Beispiel für einen möglichen Ester ist ein Salicylat in mindestens einer und möglicherweise einigen geeigneten Positionen an der Verbindung. Dieses eröffnet die Möglichkeit einer Kombinationstherapie unter Verwendung eines Indandimers und Aspirin in einem einzigen Molekül. Das Gewichtsverhältnis des Basisindandimers zu Aspirin kann so gewählt sein, dass ein Salicylat an mehreren gewählten Positionen am Dimer zur Verfügung gestellt wird.
- Es sollte selbstverständlich sein, dass die meisten Verbindungen ein oder mehrere chirale Zentren aufweisen und demzufolge als Enantiomerenpaar oder als Diastereomerengemisch vorliegen. Das kann einen Effekt auf die pharmakologischen Eigenschaften haben. Beispielsweise ist das 3C8 oben eine Mischung aus Enantiomeren. Das 3C9 ist ebenfalls eine Mischung aus Enantiomeren. Die beiden Enantiomere in 3C8 sind Diastereomere der beiden Enantiomere in 3C9. Wie anhand der Daten unten gezeigt ist, sind die beiden Enantiomere in 3C8 wahrscheinlich pharmakologisch aktiver als die beiden Enantiomere in 3C9. Tatsächlich kann eines der Enantiomere in 3C8 pharmakologisch aktiver als das andere sein.
- Pharmakologie
- Einführung
- Die erfindungsgemäßen Indandimere haben eine starke Mastzellenstabilisierungsaktivität, eine Relaxansaktivität für glatte Muskeln und eine entzündungshemmende Aktivität. Die Verbindungen sind deshalb potentielle anti-asthmatische Mittel mit Bronchodilatoraktivität. Die Mastzellenstabilisierungsaktivität der Verbindungen legen ihre potentielle Anwendung bei der Behandlung von allergischer Rhinititis, allergischer Konjunktivitis und anderen anaphylaktischen oder allergischen Erkrankungen nahe. Die enzündungshemmende Aktivität kann Anwendungen bei Gicht, rheumatischen Erkrankungen, ankylosierter Spondylitis, Polymyalgia rheumatika, temporärer Arteritis, Polyarteritis nodosa, Polymyositis und systemischer Lupus arteriosis und anderen entzündlichen Erkrankungen aufweisen. Topische Anwendungen können umfassen: atopische Ekzeme, „weeping" Ekzeme, Exema psoriasis, chronische Discoid lupus erythematosus, Flechte simplex chronicus, hypertrophischer Lichen planus, Palmare plantare pustulosis. Sie können ebenfalls ein Potential bei der Behandlung einiger maligner Erkrankungen und als Immunsuppressoren aufweisen.
- Die Relaxanzaktivität für glatte Muskeln der Verbindungen kann ein Potential bei der Behandlung von Bluthochdruck und artheriosklerotischer Gefäßerkrankungen, wie das intermittierende Hinken beim Reynaud's-Syndrom und anderen Herz- und Gefäßerkrankungen, wie die kongestive Herzinsuffizienz, Angina pektoris, zerebrale Gefäßerkrankungen und Lungenhochdruck, aufweisen. Diese Verbindungen sind ebenfalls für eine potentielle Anwendung bei der Behandlung gewisser Erkrankungen des Verdauungsextraktes, wie divertikuläre Erkrankungen und Reizcolon, indiziert sein. In ähnlicher Weise können diese Verbindungen ein Potential als Mittel für die Behandlungen von Erkrankungen des Genital-Harn-Trakts, wie verfrühte Wehen, Inkontinenz, Nierenkoliken und Erkrankungen, die mit dem Durchgang von Nierensteinen zusammenhängen, aufweisen. Mitglieder dieser Gruppe von Verbindungen können ebenfalls ein Potential als Diuretika, Analgetika, Antipyretika, lokale Anästhetika, Beruhigungsmittel des zentralen Nervensystems und Hypoglycämica aufweisen.
- Die Verbindungen wurden auf ihre Fähigkeit, die Mastzellmembranen in vitro zu stabilisieren, untersucht. Es wurden Mastzellen, die mit den Verbindungen behandelt waren und nichtbe handelte Mastzellen dahingehend stimuliert, dass sie Histamin freigeben. Eine Reduktion der Histaminfreigabe durch die behandelten Zellen im Vergleich zu den nichtbehandelten Zellen zeigt eine Stabilisierung der Membran an. Die Verbindungen wurden ihre Fähigkeit, glatte Muskeln in vitro zu relaxieren, untersucht. Der glatte Muskel wurde unter Verwendung von Calciumchlorid zur Kontraktion stimuliert und anschließend mit den Verbindungen behandelt, und die Relaxation der Kontraktion wurde für jede Verbindung gemessen. Die Verbindungen, die die meiste Aktivität in diesen Untersuchungen zeigten, wurden auf ihre Mutagenizität unter Verwendung des Salmonellenmutagenizitättests (Platteninkorporationassay) getestet. Eine von diesen (3C8) wurde weiter unter Anwendung eines in vivo Asthmamodells untersucht. Sensibilisierte Ratten wurden mit dem Wirkstoff durch Aerosol vor dem Angriff mit dem Allergen behandelt, und die Veränderungen der Atmung wurden aufgenommen. Als Ergebnis dieser Studie wurden weitere Tests durchgeführt, um die entzündungshemmende Aktivität von 3C8 zu bestimmen. Im Rattenpfotenödemtest wurde der Wirkstoff systemisch vor der Entzündungsinduktion durch Injektion von Carageenan unterhalb der planaren Sehnenhaut der Hinterpfote verabreicht. Das Volumen der Pfote wurde vor und nach der Behandlung als Index für ein Ödem bestimmt. Beim Ohrödemtest bei der Maus wurde der Wirkstoff topisch vor der Induktion der Entzündung durch topische Anwendung von Arachidonsäure verabreicht. Die Breite des Ohrs wurde vor und nach der Behandlung als Index für ein Ödem bestimmt. Die Fähigkeit von 3C8, ein Ödem zu verhindern, wurde bestimmt.
- Es folgen Protokolle von jedem dieser Assays und eine Zusammenfassung der Ergebnisse. Abkürzungen
BSS Puffersalzlösung CaCl2 Calciumchlorid CO2 Kohlenstoffdioxid DMSO Dimethylsufoxid DSCG Dinatriumcromoglycat dH20 distilliertes Wasser HCl Chlorwasserstoffsäure HEPES N-2-Hydroxyethylpiperazin-N-2-ethansulphonsäure KCl Kaliumchlorid λem Emissionswellenlänge λ2ex Anregungswellenlänge Mol Molar MgCl2 Magnesiumchlorid Min Minuten mMol Millimolar NaCl Natriumchlorid NaHCO3 Natriumhydrogencarbonat NaH2PO Natriumhydrogenphosphat NaOH Natriumhydroxid O2 Sauerstoff OPT o-Phthaldialdehyd S. E. M. mittlerer Standfehler w/v Gewicht pro Volumen v/v Volumen pro Volumen - Methoden
- Histaminfreisetzungsassay
- Es wurde vorher die Puffersalzlösung (BSS) hergestellt (NaCl 137 mMol; KCl 2,7 mMol; MgCl2 1,0 mMol; CaCl2 0,5 mMol; NaHP2O4 0,4 mMol; Glucose 5,6 mMol; HEPES 10 mMol). Dieses wurde in Teströhrchen verteilt und auf 37°C erhitzt, wobei jedes Teströhrchen 4,5 ml BSS enthielt. Die Lösungsmittelblindprobe wurde mit 0,5% (v/v) Dimethylsulfoxid (DMSO) oder 0,5% (v/v) destilliertem Wasser (dH2O) ergänzt. Die beiden positiven Kontrollen wurden mit 0,5% (v/v) dH2O/2 × 10–5 Mol Dinatriumcromoglycat (DSCG) und 0,5% (v/v) DMSO/2 × 10–5 Mol DSCG ergänzt. Die Teströhrchen für die Inkubation mit den Verbindungen enthielten 2 × 10–5 Mol Testverbindung/0,5% (v/v) DMSO. Die Inkubationsröhrchen für die Basisfreisetzung, maximale Freisetzung und den Gesamthistamingehalt enthielten keine Zugaben.
- Weibliche Wistar-Ratten (200–300 g) wurden in einer Atmosphäre gesättigtem CO2 getötet. Es wurde vorgewärmte BSS (10 ml) i. p. injiziert, und der Bauch wurde für 3 Minuten massiert. Die BSS mit den suspendierten Mastzellen und anderen Zellen wurde dem Mittellinieneinschnitt folgend angesaugt. Das Aspirat wurde für 5 Minuten bei 400 g zentrifugiert und der Überstand entfernt. Die Zellen wurden in BSS bei 4°C resuspentiert und wie zuvor zentrifugiert. Die Zellen wurden auf diese Weise insgesamt 3 Mal gewaschen. Nach dem letzten Waschen wurden die pelletisierten Zellen bei 4°C gelagert, um sie dann sobald als möglich zu verwenden.
- Die Zellen wurden in 7 ml BSS resuspendiert. Davon wurden 0,5 ml Aliquots in jede der Inkubationsröhrchen übertragen. Nach 10 Minuten bei 37°C unter leichtem Rühren wurde die Verbindung 48/80 bis zu einer Endkonzentration von 2 mg/ml gegeben, um die Histaminfreisetzung zu stimulieren. Die Zellstimulierung wurde nach 2 Minuten durch die Zugabe von 0,5 ml eiskalter BSS gestoppt, die Inkubationsröhrchen wurden dann in ein Eisbad überführt. Die Zellsuspensionen wurden für 5 Minuten bei 400 g zentrifugiert. Das Röhrchen mit dem "Gesamthistamingehalt" wurde bei 100°C für 2 Minuten vor der Zentrifugation stehengelassen. Die Überstände wurden für den Histaminassay genommen.
- Zu den 2 ml Überstand aus jedem Röhrchen wurden 0,4 ml 1 Mol NaOH und 0,1 ml oPT (1% (w/v) in Methanol gegeben. Dieses wurde bei Raumtemperatur für etwa 4 Minuten inkubiert. Die Reaktion wurde durch die Zugabe von 0,2 ml 3 Mol HCl gestoppt. Der Überstand aus jedem Inkubationsröhrchen wurde zwei Mal untersucht und gleichzeitig mit einer Standardkurve im Bereich von 0–1.000 ng/ml laufen gelassen. Die Gegenwart des fluoreszierenden Produkts der Reaktion wurde mit einem Shimadzu RF-1501 Spektrofluorophotometer, das bei λex = 360 nm, λem = 450 nm eingestellt war, gemessen.
- Jeder Wirkstoff wurde bei mindestens fünf Tieren (n = 5) getestet. Die Ergebnisse wurden als die Prozentzahl der maximalen Inhibierung der von der Verbindung 48/80 induzierten Histaminfreisetzung in der Lösungsmittelblindprobe ausgedrückt. Jeder Wirkstoff wurde gegenüber DSCG auf den gleichen Geweben verglichen. Die Basishistaminfreisetzung in den nicht behandelten Fällen wurde aufgenommen und als Prozentzahl des Gesamthistamingehalts in Suspension ausgedrückt. Das durch die Zellen freigesetzte maximale Histamin als Antwort auf die Verbindung 48/80 wurde in der relevanten Lösungsmittelblindprobe in der gleichen Weise ausgedrückt. Überall betrug die mittlere Basisfreisetzung 9,60% (S. E. M. = 1,02) des Gesamthistamingehalts der Zellen (n = 55). Die maximal stimulierte Histaminfreisetzung betrug 67,38% (S. E. M. = 2,90) in Gegenwart von 0,5% (v/v) dH2O und 54,87% (S. E. M. = 2,69) in Gegenwart von 0,5% (v/v) DMSO des Gesamthistamingehalts der Zellen (n = 55).
- Wirkungen auf glatte Muskeln
- Guinea-Schweine (350 g etwa) von jedem Geschlecht wurden in einer Atmosphäre aus gesättigtem CO2 getötet. Der Bauch wurde mit einem Mittellinieneinschnitt geöffnet, und der Dünndarm wurde entfernt.
- Es wurden Ileumsegmmente (1–1,5 cm) in Krebs-Puffer mit hohem Kaliumgehalt und ohne Calcium (NaCl 160,4 mMol; KCl 45 mMol; MgCl2 0,54 mMol; NaHP2O4 0,89 mMol; NaH2CO3 24,9 mMol; Glucose 11,1 mMol) suspendiert. Dieses wurde bei 37°C in einem Außenwandorganbad gehalten und mit 95% O2 und 5% CO2 begast. Die Gewebe wurden mit einem Faden am Boden des Organbads verankert und von Kraftstellungsgebern unter einer Ruhespannung von etwa 1 g suspendiert. Es wurden die isotonischen Kontraktionen unter Verwendung eines MacLab/4e-Systems zusammen mit dem Chart 3.3.1 Softwarepackage aufgenommen. Überflüssiges Gewebe wurde bei 4°C in Krebs-Puffer (NaCl 236,5 mMol; KCl 4,7 mMol; CaCl2 2,5 mMol; MgCl2 0,54 mMol; NaHP2O4 0,89 mMol; NaHCO3 24,9 mMol; Glukose 11,1 mMol) für maximal 48 Stunden aufbewahrt.
- Es wurden vier Gewebesegmente suspendiert und gleichzeitig beobachtet. Die Kontraktionen wurden durch Zugabe von 25 μl 1 Mol CaCl2 (Endkonzentration von 2,5 mMol) initiiert. Die Kontrationen stabilisierten sich mit der Zeit, 10–15 Minuten und konnten bis zu 45 Minuten von der Zugabe des CaCl2 an aufrechterhalten werden.
- Vorratslösungen mit dem Wirkstoff wurden zu 10–3 Mol in 50 (v/v) DMSO hergestellt. Diese wurden verdünnt, um 10–4 Mol in 5 (v/v) DMSO und 10–5 Mol in 0,5% (v/v) DMSO herzustellen. Bei Fällen geringer Löslichkeit wurde der 10–3 Mol Vorrat in höheren Konzentrationen von DMSO hergestellt. Lösungsmittel"blindproben" lösungen wurden hergestellt, die 50%, 5% und 0,5% (v/v) DMSO (oder entsprechend geeignet) hielten. Die Wirkstofflösung wurde in das Organbad gegeben, so bald eine stabile Kontraktion des Gewebes erreicht worden war. Ein Antwortassay bei der Gesamtdosis wurde im Bereich von 5 × 10–8 Mol bis 10–5 Mol durchgeführt. Das Organbad wurde ausgewaschen und man ließ das Gewebe relaxieren. Ein zweiter Antwortassay mit der Gesamtdosis wurde durchgeführt, wobei nur die DMSO-"Blindproben" -Lösungen verwendet wurden.
- Jeder Wirkstoff wurde zweimal bei mindestens drei verschiedenen Tieren (n = 3) getestet. Die Ergebnisse wurden als Prozentzahl der Inhibierung der durch CaCl2 induzierten Kontraktion für jedes Gewebe bei jeder Konzentration des Wirkstoffs in DMSO ausgedrückt. Der Effekt von DMSO für jedes Gewebe bei jeder Konzentration wurde von dem Effekt des Wirkstoffs in DMSO abgezogen, womit man den Effekt des Wirkstoffs allein erhielt. Eine log-Kurve Dosis gegen Antwort wurde für jeden Wirkstoff unter Verwendung des Mittels und des Standardfehlers des Mittels für die gesamten Ergebnisse aufgetragen.
- Salmonellen-Mutageniziätsest
- Die Verbindungen wurden auf ihre Mutagenizität nach einem Protoll von Ames et al. (Mutation Res. 31, 347–364, 1975), das von Maron and Ames (Mutation Res. 113, 173–215, 1983) modifiziert worden ist, getestet. Es wurden die Histidin benötigenden Stämme TA98, TA100, TA102 und TA1535 von Salmonella Typhimurium LT2 für den Mutagenizitätstest verwendet. Diese Stämme enthalten eine Anzahl anderer Mutationen, die ihre Fähigkeit stark verbessern, Mutagene nachzuweisen. Diese sind (1) eine Mutation (rfa+), die einen Teilverlust der Lipopolysaccharidzellwand verursacht und somit das Permeationsvermögen der Zellen gegenüber größeren Molekülen erhöht, und (2) eine Deletion (uvrB), die den Verlust des DNA-Excisionsreparatursystems verursacht. Beim TA102 ist das Excisionsreparatursystem (uvrB+) erhalten, womit dieser in der Lage ist, Mutagene, die dieses System brauchen, nachzuweisen. Außerdem enthält TA102 das PAQ1-Plasmid mit der his G428 Mutation, was diesem Stamm eine Tetracyclinresistenz verleiht. TA98, TA100 und TA102 weisen ebenfalls ein R-Faktor-Plasmid, PKM101 auf, das ein Ampicillinresistenzgen enthält. Die Mutation des Genoms, was eine Umkehrung der Histidinunabhängigkeit verursacht, kann unter Verwendung von Selektionsmedien nachgewiesen werden.
- Lösungen aus Testverbindungen wurden in DMSO im Bereich von 0– 50 mg/ml hergestellt. Topagar (2 ml), der bei oder oberhalb 45°C gehalten wird und 0,5 mMol L-Histidin HCl/0,5 mMol Biotin enthält, wurde in sterile 5 ml Probenröhrchen verteilt. Es wurde eine frische Übernachtkultur des relevanten Stamms (0,1 ml) zusammen mit der Testverbindung (0,1 ml) hinzugegeben werden. Es können ebenfalls Lebermikrosomenenzyme der Ratte enthalten sein (0,5 ml eines S9-Mix), um auf mutagene Metaboliten der Testverbindungen zu testen. Der Inhalt der Probenröhrchen wurde auf Minimalglukoseagarplatten übertragen und für eine Stunde zum Trocknen stehen gelassen. Die Platten wurden umgedreht und bei 37°C für 48 h inkubiert. Das Zellenwachstum findet nur bei einem austretenden Mutationsereignis statt. Die Anzahl der revertanten Kolonien auf jeder Platte wurde gezählt. Sechs Negativkontrollplatten, einschließlich DMSO und sechs Positivkontrollplatten, einschließlich ein diagnostisches Mutagen, wurden zusammen mit jeder Verbindung getestet. Drei bis vier Testplatten wurden für jede Verbindung bei jeder der fünf verschiedenen Konzentrationen verwendet. Ein signifikanter Anstieg der Anzahl von revertanten Kolonien auf einer Testplatte im Vergleich zur Hintergrundinversionsrate könnte anzeigen, dass die Testverbindung mutagen war. Ein Signifikantstest wurde durchgeführt, wobei der multiple Vergleichstest von Dunnett (Dunnett C. W., Jnl. AM. Statist. Assoc. 50, 1096–1121, 1955) angewendet wurde.
- In vivo Bronchialasthma-Modell
- Es wurden Experimente mit männlichen Wistar-Ratten im Alter von 10–12 Wochen (200–250 g) durchgeführt. Die Sensibilisierung erfolgte durch die Injektion (1 ml s. c.) von Ovalbumin (1 mg/ml)/Aluminiumhydroxid (200 mg/ml) und Freund'schem kompletten Adjuvants (1 ml i. p.).
- Drei Wochen nach der Sensibilisierung wurde jedes Tier mit Natriumphenobarbiton (40 mg/kg i. p.) sediert, und die Sedierung wurde mit zusätzlichen Injektionen (5 mg/kg i. p.), falls erforderlich, aufrechterhalten. Die Nase wurde mit einem chirurgischen Band verschlossen, um die Ablagerung von Aerosolen zu verhindern. Das Tier wurde in eine Atmungskammer gesetzt, und es wurden die Atmungsparameter mit einem Differenzialvolumenenergieumwandler gemessen. Die Tiere wurden mit einem Ethanol-Aerosol (50% v/v als Negativkontrolle), DSCG (5 mg/ml in Ethanol, 50% v/v als Positivkontrolle) oder 3C8 (5 mg/ml in Ethanol 50% v/v) behandelt. Die Tiere wurden danach mit einem Salzlösungsaerosol (Negativkontrolle oder Ovalbumin 5% w/w) stimuliert. Änderungen in der Atmung wurden für 3 Stunden beobachtet. Jedes Tier bekam drei weitere Behandlungen, um eine Bronchialhyperreaktivität zu induzieren, eine Bedingung, die sehr stark dem Bronchialasthma ähnelt.
- Drei bis vier Wochen nach der Anfangsbehandlung wurde das Experiment wiederholt. Die Tiere wurden einem Aerosol aus Acetylmethylcholin (Metacholin) bei einer Dosis (8 mg/ml) ausgesetzt, das eine signifikante Antwort nur in hyperreaktiven Luftwegen stimuliert. Keines der Tiere wurde mit dem Wirkstoff vorbehandelt. Änderungen von Antworten der Luftwege wurden für 1 Stunde beobachtet.
- In vivo Entzündungsmodelle
- Es wurde das Rattenpfotenödemmodell unter Verwendung von weiblichen Wistar-Ratten (180–200 g) durchgeführt. Die Tiere wurden mit Natriumpentobarbiton sediert, 40–70 mg/kg i. p. Die Tiere wurden durch eine i. p.-Injektion in einem Konzentrationsbereich des Testwirkstoffs (0–100 mg/kg) in 50% DMSO) oder Hydrokortison (100 mg/kg in 50% DMSO) oder Indomethancin (100 mg/mk in 50% DMSO) behandelt. Nach 30 Minuten wurden die Ödeme durch Injektion von Carageenan (100 μl bei 2% w/v) unterhalb der plantaren Sohlenplatte der Hinterpfote induziert. Das Volumen der Pfote wurde gemessen, sowohl vor und 60 Minuten nach der Behandlung, durch die Verdrängung von Wasser in einem Messzylinder. Das Pfotenödem wurde berechnet, indem das Pfotenvolumen vor und nach der Induktion des Ödems verglichen wurde, und es wurde als Prozentzahl normal ausgedrückt.
- Das Ohrödemmodell bei der Maus wurde unter Verwendung von Laca-Mäusen (25–35 g) von jedem Geschlecht durchgeführt. Die Tiere wurden mit Fentanyl/Fluanizon (Hypnorm, Janssen) sediert. Ein Ohr wurde durch die topische Anwendung einer der Testverbindungen, Indomethacin oder Dexamethazon (alle zu 300 μg Ohr in Aceton)-Wirkstoff, behandelt. Nach 30 Minuten wurde das Ödem durch die topische Anwendung von Arachidonsäure (10 μl bei 0,4 g/ml in Aceton). Die Dicke von jedem Ohr wurde gemessen, sowohl vor und 60 Minuten nach der Induktion des Ödems, wobei eine Mikrometerschraubenlehre verbunden wurde.
- Das Ohrödem wurde berechnet, indem die Ohrbreite vor und nach der Induktion des Ödems verglichen wurde, und es wurde als Prozentzahl normal ausgedrückt.
- Ergebnisse Mastzellensensibilisierung und Relaxation der glatten Muskeln
- Die Ergebnisse der Assays über die Histaminfreisetzung und den Effekt auf den glatten Muskel sind in den anliegenden Ergebnistabellen zusammengefasst. Die Ergebnisse von einigen Verbindungen sind in den anliegenden Graphen erläutert. Diese Ergebnisse zeigen, dass diese Verbindungen eine große Vielzahl von Wirkungen bei der Relaxierung des glatten Muskels und der Mastzellenstabilisierung zeigen, und dass diese beiden Effekte nicht miteinander verwandt sind (das heißt, ein guter Mastzellenstabilisator ist nicht notwendigerweise ein gutes Relaxans für den glatten Muskel und umgekehrt).
- Ergebnisse des Histamin-Freisetzungstests und der glatten Muskeln

-

-

-

-

- Toxikologie
- Gemäß dem Salmonellen-Mutagenizitätstest erhöhten 1C13, 1C14, 1C125/26 und 3C8 nicht signifikant die Anzahl der revertanten Kolonien in den Salmonella typhimurium LT2-Stämmen TA98, TA100, TA102 und TA1535 bei Konzentrationen bis zu 5 mg/Platte in Gegenwart oder Abwesenheit von Lebermikrosomenenzymen (S9-Mix). Die Verbindung 3C9 erhöhte nicht signifikant die Anzahl der revertanten Kolonien in dem Salmonella typhimurium LT2-Stamm, TA98, inkubiert ohne den S9-Mix, bei Konzentrationen bis zu 5 mg/Platte. Daraus ist geschlossen worden, dass 1013, 1C14, 1C25/26, 3C8 und 3C9 nicht mutagen sind, wobei die oben erwähnten Konzentrationen und Bakterienstämme, nach dem Salmonellen-Mutagenizitätstest nach Ames, verwendet wurden.
- Bronchialasthma-Modell
- Alle Tiere antworteten auf die Aerosolbehandlung mit einem 30 %igen Abfall der Atmungsrate und einem 50%igen Abfall des Atemvolumens, ungeachtet des Gehalts der Aerosollösung. Dieses kann gut die frühe Antwort der Tiere, die Ovalbumin bekamen, maskiert haben, weil keine beobachtet wurde. Die Tiere wurden in vier Behandlungsgruppen aufgeteilt:
- Gruppe I – Negative Kontrolle
- Diese Tiere wurden mit Ethanol (50% v/v) durch Aerosol 30 Minuten vor der Salzlösungsbehandlung (NaCl 0,9% w/v) ebenfalls durch Aerosol, behandelt. Es gab einen Abfall der Atmungsrate von 30% und des Atemvolumens von 50% bei der Antwort auf das Aerosol. Beide Parameter kamen innerhalb 10 Minuten auf Normal zurück und blieben über den Beobachtungszeitraum (
1 ) normal. Nach der Metacholinbehandlung fiel das Atemvolumen um 50 %, die Atmungsrate stieg allerdings um 10% (2 ). Dieses kann eine Antwort auf das Metacholin sein, die nicht ausreicht, um die erwartete Antwort auf das Aerosol zu überwinden. Eine Antwort auf Metacholin war nicht erwartet, da die Tiere nicht mit einem Allergen behandelt waren. - Die Post-mortem-Untersuchung zeigte, dass die Tiere alle eine schwere sterile Peritonitis entwickelt hatten. Dieses nahm die Form einer sehr starken, vaskularisierten Vibrose innerhalb des Abdomens, insbesondere um die Leber und die oberen Därme, an. Außerdem entwickelten sich kleine käseartige Knötchen, wieder primär um die Leber, allerdings ebenfalls über den Bauch mit einer gelegentlichen Konzentration an den Injektionsstellen. Es wird angenommen, dass diese Entwicklung ein Ergebnis oder vielen i. p.-Phenobarbitoninjektionen, ein saures Irritationsmittel, ist.
- Gruppe II – Positive Kontrolle
- Diese Tiere wurden Ovalbumin (5% w/v) durch Aerosol ausgesetzt. Es gab einen Abfall der Atmungsrate von 35% und des Atemvolumens von 40% als Antwort auf das Aerosol. Die frühe Phase der allergischen Antwort auf das Ovalbumin wird durch den Aerosoleffekt maskiert. Beide Parameter gingen innerhalb 10 Minuten auf normal zurück und blieben für 100 Minuten normal. Zwei Stunden (120 ml) nach der Ovalbuminbehandlung stieg die Atmungsrate um 35% und das Atemvolumen stieg um 60%. Dieses repräsentiert die späte Phase der allergischen Antwort.
- Beide Parameter kamen innerhalb 30 Minuten auf normal zurück und verblieben normal für den Rest der Beobachtungsdauer (
1 ). Nach der Metacholinbehandlung fiel das Atemvolumen um 25 %, allerdings stieg die Atmungsrate um 50%. Dieses zeigt eine Antwort auf das Metacholin an, die ausreicht, um die erwartete Antwort auf das Aerosol zu überwinden, was impliziert, dass mehrere Allergenbehandlungen (Ovalbumin) eine Hyperreaktivität im Luftweg induziert haben. Beide Parameter kamen auf normal in 10 Minuten zurück und verblieben während der Beobachtungsdauer (2 ) normal. - Die Post-mortem-Untersuchung zeigte, dass die Tiere alle eine schwere sterile Peritonitis entwickelt hatten. Die Natur und Schwere der Reaktion war ähnlich derjenigen, die bei den Tieren der Gruppe I beobachtet wurde.
- Gruppe III – Behandlungskontrolle
- Diese Tiere wurden Dinatriumcromoglycat (5 mg/ml in Ethanol, 50% v/v) und 30 Minuten später Ovalbumin (5% v/w) durch Aerosol ausgesetzt. Es gab einen Abfall der Atmungsrate von 30 und des Atemvolumens von 40% als Antwort auf das Aerosol. Beide Parameter kamen innerhalb von 10 Minuten auf normal zurück, das Atemvolumen blieb über den Beobachtungszeitraum normal. Die Atmungsrate stieg um 10% 90 Minuten nach der Ovalbuminbehandlung, was etwa 15 Minuten dauerte. Der Hauptpeak der späten Phase trat bei keinem der Parameter auf, was anzeigt, dass die Dinatriumcromoglycat-Vorbehandlung die Tiere davor schützt, eine Asthmaantwort auf das Ovalbumin (
1 ) zu entwickeln. Nach der Metacholinbehandlung fiel das Atemvolumen um 25%, allerdings stieg die Atmungsrate um 5%. Dieses kann ei ne Antwort auf das Metacholin anzeigen, die nicht ausreicht, die erwartete Antwort auf das Aerosol zu überwinden (2 ). Die reduzierte Antwort auf Metacholin, verglichen mit den Kontrollen der Gruppe II, zeigt an, dass die Dinatriumchromoglycatbehandlung die Tiere davor schützt, eine Hyperreaktivität in den Luftwegen induzieren. - Die Post-mortem-Untersuchung zeigte, dass die Tiere alle eine schwere sterile Peritonitis entwickelt hatten. Die Natur der Reaktion war ähnlich derjenigen, die bei den Tieren der Gruppe I und Gruppe II beobachtet wurden. Die Reaktion war weniger stark, was durch die Abwesenheit der Vaskularisierung des fibrotischen Gewebes angezeigt wird, und was nahe legt, dass das Dinatriumcromoglycat in der Weise gewirkt haben kann, diese Tiere zu schützen.
- Gruppe IV – Behandlungstest
- Diese Tiere wurden 3C8 (5 mg/ml in Ethanol, 50% v/v) und 30 Minuten später Ovalbumin (5% w/v) durch Aerosol ausgesetzt. Es gab einen Abfall der Atmungsrate von 40% und des Atemvolumens von 60% als Antwort auf das Aerosol. Beide Parameter kamen auf 90% normal innerhalb 10 Minuten zurück und schwankten um 90% normal für den Rest des Beobachtungszeitraums. Der Hauptpeak in der letzten Phase kam bei keinem der Parameter vor, was anzeigt, dass die 3C8-Vorbehandlung die Tiere davor geschützt hat, eine Asthmaantwort auf das Ovalbumin (
1 ) zu entwickeln. Nach der Metacholinbehandlung fiel das Atemvolumen um 40%, allerdings stieg die Atmungsrate um 10%. Dieses kann eine Antwort auf das Metacholin anzeigen, die nicht ausreicht, um die erwartete Antwort auf das Aerosol (2 ) zu überwinden. Die verminderte Antwort auf das Metacholin, im Vergleich zu den Kontrollen der Gruppe II, zeigt an, dass die Behandlung mit 3C8 die Tiere davor geschützt hat, eine Hyperreaktivität in den Luftwegen zu induzieren. - Die Post-mortem-Untersuchung zeigte, dass keines der Tiere die sterile Peritonitis, die in den anderen Gruppen beobachtet wurde, entwickelt hatte. Dieses könnte anzeigen, dass das 3C8 dahingehend gewirkt hat, diese Tiere vor einer Entzündungsantwort zu schützen.
- Schlussfolgerung
- Das Protokoll, das dafür verwendet wurde, allergisches Asthma und eine Bronchialhyperreaktivität in männlichen Wistar-Ratten zu induzieren, war erfolgreich, mit Ausnahme bei der Beobachtung der frühen Phase der allergischen Reaktionen. Die Behandlungskontrolle (Dinatriumcromoglycat) war insoweit erfolgreich, dass sie die messbaren Antworten auf das Allergen blockierte und die Entwicklung von Bronchialhyperreaktivität verhinderte. Die Testverbindung 3C8 war ebenfalls erfolgreich, wenn nicht gar marginal besser, bei der Blockierung der allergischen Antwort und der Verhinderung der Entwicklung der Bronchialhyperreaktivität. Außerdem kann 3C8 als entzündungshemmendes Mittel fungieren, was durch die vollständige Abwesenheit von Peritonitis in den mit 3C8 behandelten Tieren angezeigt wird.
- Entzündungsmodelle
- Ödemmodell an der Rattenpfote
- Ein Bereich von Dosen von 3C8, im Vergleich zur Antwort auf Einzeldosen Indomethacin und Hydrocortison, ist wie folgt:
-

- Ödemmodell am Ohr der Maus
- Antworten des Mausohrs auf Einzeldosen einer Anzahl von Verbindungen im Vergleich zur Antwort auf Indomethacin und Dexamethanson, jeweils bei einer Dosis von 300 μg pro Ohr, topisch verabreicht 30 Min vor der Verabreichung von Arachidonsäure. Die Werte werden als Prozentzahl des Anstiegs der Ohrdicke eine Stunde nach Verabreichung der Arachidonsäure (alle n = 4, außer 8C4 (n = 5)) und der Lösungsmittelkontrollen (n = 8) aufgedrückt. Die Ergebnisse legen nahe, dass die entzündungshemmende Aktivität nicht mit der Mastzellenstabilisierungsaktivität zusammenhängt.
-

- Es sollte selbstverständlich sein, dass die Verbindungen geeignete pharmakologische Eigenschaften, die anders als die oben beschriebenen sind, aufweisen können. Die Erfindung ist nicht auf die vorliegend beschriebenen Ausführungsformen beschränkt, die jeweils im Detail verändert werden können. ANHANG 1 Liste der versendeten Abkürzungen
AlCl3 Aluminiumchlorid aq wässrig b. p. Schmelzpunkt BrCH2C6H4CO2CH3 Methyl 4-(brommethyl)benzoat BrCH2CO2CH3 Brommethylacetat BSS Puffersalzlösung CaCl2 Calciumchlorid C2H5I Jodethan C6H3(CH3)Br(CH3) Brom-m-xylol C6H5CH2Br Benzylbromid CDCl3 Chloroform-d CF3SO3Si(CH3)3 Trimethylsilyltrifluormethansulfonat (TMS-Triflate) CH(OCH3)3 Trimethylsilylorthoformat CH3C6H4SO3H·H2O p-Toluolsulfonsäure CH3I Jodmethan ClCH2CH2COCl β-Chlorpropionylchlorid CO2 Kohlendioxid CS2 Kohlenstoffdisulfid [(C6H5)3P]3RhCl Tris(triphenylphosphin)rhodium(1)chlorid (Wilkinson-Katalysator) [(CH3)3CO]3Al Aluminium-tri-tert.-butoxid DCM Dichlormethan dH2O destilliertes Wasser DMSO Dimethylsulfoxid DSCG Dinatriumcromoglycat Et2O Ether Et3N Triethylamin EtOAc Ethylacetat EtOH Ethanol H2C=CHCH2Br Allylbromid H2NNH2·H2O Hydrazinhydrat·Monohydrat H2O Wasser H2SO4 Schwefelsäure HCl Chlorwasserstoffsäure HEPES N-2-Hydroxyethylpiperazin-N-2-ethansulfonsäure HOCH2CH2OH Ethylenglykol IR Infrarot KCl Kaliumchlorid LDA Lithiumdiisopropylamid Mol Molar MgCl2 Magnesiumchlorid Min Minuten μl Mikroliter mMol Millimolar m. p. Schmelzpunkt N2 Stickstoff NaBH4 Natriumborhydrid NaCl Natriumchlorid NaCN(BH3) Natriumcyanoborhydrid NaHCO3 Natriumhydrogencarbonat NaHCO3 Natriumbicarbonat NaH2PO Natriumhydrogenphosphat NaOH Natriumhydroxid Na2SO4 Natriumsulfat NH4Cl Ammoniumchlorid NMR Kernmagnetresonanz O2 Sauerstoff OPT o-Phthaldialdehyd Pd Palladium RT Raumtemperatur tBuOH tert.-Butanol tBuOK Kalium-tert.-butoxid S. E. M. mittlerer Standardfehler THF Tetrahydrofuran TLC Dünnschichtchromatographie μl Mikroliter Triflic Acid Trifluormethansulfonsäure TMS Triflate Trimethylsilyltrifluormethansulfonat v/v Volumen pro Volumen w/v Gewicht pro Volumen ZnI2 Zinkjodid λem Emissionswellenlänge λ2ex Anregungswellenlänge 1C4 2-(I-Indanyl)-2-methylindan 1C5 2-(1-Ind-1-enyl)-2-methylindan-1-ol 1C7 1-(2-(2-Methylindanyl))ind-1-en 1C8 (2-(1-Ind-1-enyl)-2-methyl-1-acetoxyindan 1C9 2-(3,3-Dimetyl-1-ind-1-enyl)-2-methylindan-1-on 1C10 2-(1-Ind-1-enyl)-2-ethylindan-1-on 1C12 2-(1-Ind-1-enyl)-2-prop-2-enylindan-1-on 1C13 2-(1-Ind-1-enyl)-2-propylindan-1-on 1C15 2-(1-Ind-1-enyl)-2-prop-2-enylindan-1-ol 1C16 2-(1-Ind-1-enyl)-2-prop-2-enyl-1-acetoxyindan 1C17 2-(1-Ind-1-enyl)-2-propylindan-1-ol 1C18 2-(1-Ind-1-enyl)-2-propylindan-1-ol 1C19 2-(Ind-1-enyl)-2-propanyl-1-acetoxyindan 1C20 2-(1-Ind-1-enyl)-2-pent-2-enylindan-1-on 1C21 2-(1-Ind-1-enyl)-2-pentylindan-1-on 1C22 2-(1-Ind-1-enyl)-2-pent-2-enylindan-1-ol 1C23 2-(1-Ind-1-enyl)-2-pent-2-enylindan-1-ol 1C24 2-(1-Ind-1-enyl)-2-benzylindan-1-on 1C25 und 1C26 2-(1-Ind-1-enyl)-2-benzylindan-1-ol 1C27 2-(1-Ind-1-enyl)-2-benzyl-1-acetoxyindan 1C31 2-(1-Ind-1-enyl)-2-p-methoxycarbonylphenylmethyl-indan-1-on 1C32 2-(1-Ind-1-enyl)-2-p-carboxyphenylmethylindan-1-on 1C33 2-(1-Ind-1-enyl)-2-methoxycarbonylmethylindan-1-on 1C34 2-(1-Indenyl)-2-carboxymethylindan-1-on 1C35 2-(1-Ind-1-enyl)-2-natriumoxycarbonylmethylindan-1-on IC38 2-(1-Ind-1-enyl)-2-acetoxymethylindan-1-on 2C3 1-(2-Indenyl)-1-ethylindan-2-on 2C4 1-(2-Indenyl)-indan-2-on 2C6 1-(2-Indenyl)-1-prop-2-enylindan-2-on 2C7 1-(2-Indenyl)-1-benzylindan-2-on 2C8 1-(2-Indenyl)-1-benzylindan-2-ol 2C10 1-(Benzyl-2-inden-2-enyl)-inden-2-acetoxy 3C3 2-(2-Indenyl)-2-prop-2-enylindan-1-on 3C4 2-(2-Indenyl)-2-propyindan-1-on 3C5 2-(2-Indenyl)-2-benzylindan-1-on 3C6 2-(2-Indenyl)-2-benzylindan-1-ol 3C7 2-(2-Indenyl)-2-benzylindan-1-ol 3C8 2-(2-Indenyl)-2-benzylindan-1-ol 3C9 2-(2-Indenyl)-2-benzylindan-1-ol 3C10 2-(2-Indenyl)2-benzyl-1-acetoxyindan 3C11 2-(2-Indenyl)2-p-methoxycarbonylphenylmethylindan-1-on 3C13 2-(2-Indenyl)-2-p-methoxycarbonylphenylmethyl-4-methoxyindan-1-on 3C18 2-(2-Indenyl-2-benzyl-6-brom-5,7-dimethylindan-1-on 3C19 2-(2-Indenyl)-2-benzyl-6-brom-5,7-dimethylindan-1-ol 3C20 2-(2-Indenyl)-2-benzyl-6-brom-5,7-dimethylindan-1-ol 3C22 2-(2-Indenyl)-2-benzyl-5-brom-4,6-dimethylindan-1-on 3C23 2-(2-Indenyl)-2-benzyl-5-brom-4,6-dimethylindan-1-ol 3C24 2-(2-Indenyl)-2-benzyl-5-brom-4,6-dimethylindan-1-ol 3C31 2-(2-Indenyl)-2-benzyl-5,7-dimethylindan-1-on 3C32 2-(2-Indenyl)-2-benzyl-5,7-dimethylindan-1-ol 3C33 2-(2-Indenyl)-2-benzyl-5,7-dimethylindan-1-ol
Claims (24)
1 Pharmazeutische Zusammensetzung, umfassend eine Verbindung
von jedweder der folgenden Formeln:  worin
worin in
Formeln 1 und 3 R3 bis R15
in
Formel 2 R3 bis R15
ausgewählt
werden aus einem oder mehreren des Gleichen oder Unterschiedlichen
von:
H, Halo, Hydroxy, Alkoxy, Aryloxy, Acetoxy, Carboxy, Alkylcarbonyl,
Hydrocarbonyl, Amino, Amido, Alkylamino, Hydroxylamino, Aralkyl-Gruppen,
mono- und polybenzoiden Aryl-Gruppen, substituierten Aryl-Gruppen,
Alkyl, enthaltend 1 bis 10 Kohlenstoffatome oder Cycloalkyl-Gruppen,
enthaltend 3 bis 8 Kohlenstoffatome, substituiertem Alkyl oder Cycloalkyl,
worin das Alkyl oder Cycloalkyl substituiert wird durch jedwedes
eine oder mehrere des Gleichen oder Unterschiedliche von Halo, Oxo,
Hydroxy, Alkoxy, Aryloxy, Acetoxy, Carboxy, Carbonyl, Amino, Amido,
Alkylamino, Hydroxylamino, Aralkyl-Gruppen, mono- und polybenzoiden
Aryl-Gruppen, substituierten Aryl-Gruppen,
in Formeln 1 und
3 R1, 1R1 und in Formel 2 R2, 1R2 jedwedes eine
oder mehrere von Hydrocarbonyl, Alkylcarbonyl, Wasserstoff, Hydroxy
oder Acetoxy sind,
in Formel 1 jedwedes eine oder mehrere von
R1, 1R1;
R3, 1R3;
R10, 1R10 zusammen
Oxo darstellen können;
in
Formel 2 jedwedes eine oder mehrere von R2, 1R2; R3, 1R3; R14, 1R14 zusammen Oxo
darstellen können,
in
Formel 3 jedwedes eine oder mehrere von R1, 1R1; R3, 1R3; R14, 1R14 zusammen Oxo
darstellen können.
Verbindung von Formel 1 oder 2 nach Anspruch 1, ausklammernd
Folgendes
1-(2-Indenyl)-1-methyl-indan-2-on;
1-(2-Indenyl)-indan
und
2-(1-Indenyl)-indanon.
Verbindung nach Anspruch 2, worin in Formel 1 R1, 1R1;
R3, 1R3 und
R10, 1R10 nicht
jeweils Oxo darstellen.
Verbindung nach Anspruch 2 oder 3, worin in Formel
1 R3 bis R7 Wasserstoff
sind.
Verbindung nach einem der Ansprüche 2 bis 4, worin in Formel
1 R10 bis R14 Wasserstoff
sind.
Verbindung nach einem der Ansprüche 2 bis 5, worin in Formel
1 R1, 1R1 H, OH darstellt.
Verbindung nach einem der Ansprüche 2 bis 6, worin in Formel
1 R15 eine Benzyl-Gruppe darstellt.
Verbindung nach Anspruch 2, worin in Formel 2 R1, 1R2;
R3, 1R3 und
R14 1R14 nicht
jeweils Oxo darstellen.
Verbindung nach Anspruch 2 oder 8, worin in Formel
2 R3 bis R7 Wasserstoff
sind.
Verbindung nach einem der Ansprüche 2 oder 8 oder 9, worin
in Formel 2 R10 und R13 Wasserstoff
sind.
Verbindung nach einem der Ansprüche 2 oder 8 bis 10, worin
in Formel 2 R2, 1R2 H, OH darstellt.
Verbindung nach einem der Ansprüche 2 oder 8 bis 11, worin
in Formel 2 R15 eine Benzyl-Gruppe darstellt.
Verbindung der Formel 3 wie definiert nach Anspruch
1.
Verbindung nach Anspruch 13, worin R1, 1R1; R3, 1R3 und R14, 1R14 nicht
jeweils Oxo darstellen.
Verbindung nach Anspruch 13 oder 14, worin R4 bis R7 Wasserstoff
darstellen.
Verbindung nach einem der Ansprüche 13 bis 15, worin R10 bis R13 Wasserstoff
darstellen.
Verbindung nach einem der Ansprüche 13 bis 16, worin R1, 1R1 H,
OH darstellt.
Verbindung nach einem der Ansprüche 13 bis 17, worin R15 eine Benzyl-Gruppe darstellt.
Verbindung, ausgewählt aus dem Folgenden: 1C4 2-(1-Indanyl)-2-methylindan
1C5 2-(1-Ind-1-enyl)-2-methylindan-1-ol
1C7 1-(2-(2-Methylindanyl)ind-1-en
1C8 2-(1-Ind-1-enyl)-2-methyl-1-acetoxyindan
1C9 2-(3,3-Dimethyl-1-ind-1-enyl)-2-methylindan-1-on
1C10 2-(1-Ind-1-enyl)-2-ethylindan-1-on
1C12 2-(1-Ind-1-enyl)-2-prop-2-enylindan-1-on
1C13 2-(1-Ind-1-enyl)-2-propylindan-1-on
1C15 2-(1-Ind-1-enyl)-2-prop-2-enylindan-1-ol
1C16 2-(1-Ind-1-enyl)
-2-prop-2-enyl-1-acetoxyindan
1C17 2-(1-Ind-1-enyl)-2-propylindan-1-ol
1C18 2-(1-Ind-1-enyl)-2-propylindan-1-ol
1C19 2-(Ind-1-enyl)-2-propanyl-1-acetoxyindan
1C20 2-(1-Ind-1-enyl)-2-pent-2-enylindan-1-on
1C21 2-(1-Ind-1-enyl)-2-pentylindan-1-on
1C22 2-(1-Ind-1-enyl)-2-pent-2-enylindan-1-ol
1C23 2-(1-Ind-1-enyl)-2-pent-2-enylindan-1-ol
1C24 2-(1-Ind-1-enyl)-2-benzylindan-1-on
1C25
und 1C26 2-(1-Ind-1-enyl)-2-benzylindan-1-ol
1C27 2-(1-Ind-1-enyl)-2-benzyl-1-acetoxyindan
1C31 2-(1-Ind-1-enyl)-2-p-methoxycarbonylphenylmethy 1-indan-1-on
1C32 2-(1-Ind-1-enyl)-2-p-carboxyphenylmethylindan-1-on
1C33 2-(1-Ind-1-enyl)-2-methoxycarbonylmethylindan-1-on
1C34 2-(1-Indenyl)-2-carboxymethylindan-1-on
1C35 2-(1-Ind-1-enyl)-2-natrium-oxycarbonylmethylindan-1-on
1C38 2-(1-Ind-1-enyl)-2-acetoxymethylindan-1-on
2C3 1-(2-Indenyl)-1-ethylindan-2-on
2C4 1-(2-Indenyl)-indan-2-on
2C6 1-(2-Indenyl)-1-prop-2-enylindan-2-on
2C7 1-(2-Indenyl)-1-benzylindan-2-on
2C8 1-(2-Indenyl)-1-benzylindan-2-ol
2C10 1-(Benzyl-2-inden-2-enyl)-inden-2-acetoxy
3C3 2-(2-Indenyl)-2-prop-2-enylindan-1-on
3C4 2-(2-Indenyl)-2-propylindan-1-on
3C5 2-(2-Indenyl)-2-benzylindan-1-on
3C6 2-(2-Indenyl)-2-benzylindan-1-ol
3C7 2-(2-Indenyl)-2-benzylindan-1-ol
3C8 2-(2-Indenyl)-2-benzylindan-1-ol
3C9 2-(2-Indenyl)-2-benzylindan-1-ol
3C10 2-(2-Indenyl)-2-benzyl-1-acetoxyindan
3C11 2-(2-Indenyl)-2-p-methoxycarbonyl-phenylmethylindan-1-on
3C13 2-(2-Indenyl)-2-p-methoxycarbonyl-phenylmethyl-4-methoxyindan-1-on
3C18 2-(2-Indenyl-2-benzyl-6-bromo-5,7-dimethylindan-1-on
3C19 2-(2-Indenyl)-2-benzyl-6-bromo-5,7-dimethylindan-1-ol
3C20 2-(2-Indenyl)-2-benzyl-6-bromo-5,7-dimethylindan-1-ol
3C22 2-(2-Indenyl)-2-benzyl-5-bromo-4,6-dimethylindan-1-on
3C23 2-(2-Indenyl)-2-benzyl-5-bromo-4,6-dimethylindan-1-ol
3C24 2-(2-Indenyl)-2-benzyl-5-bromo-4,6-dimethylindan-1-ol
3C31 2-(2-Indenyl)-2-benzyl-5,7-dimethylindan-1-on
3C32 2-(2-Indenyl)-2-benzyl-5,7-dimethylindan-1-ol
3C33 2-(2-Indenyl)-2-benzyl-5,7-dimethylindan-1-ol
Pharmazeutische Zusammensetzung, umfassend eine
Verbindung nach einem der Ansprüche
2 bis 12 und einen pharmazeutisch zulässigen Träger.
Pharmazeutische Zusammensetzung, umfassend eine
Verbindung nach einem der Ansprüche
13 bis 19 und einen pharmazeutisch zulässigen Träger.
Verwendung einer Verbindung von Formeln 1 bis 3
wie definiert nach Anspruch 1 bei der Herstellung eines Medikamentes
zum Erreichen von Relaxationsaktivität der glatten Muskulatur und/oder
Mastzellen-Stabilisationsaktivität
und/oder antiinflammatorischer Aktivität.
Verwendung einer Verbindung von Formel 1 oder 2
nach einem der Ansprüche
2 bis 12 bei der Herstellung eines Medikamentes zum Erreichen von
Relaxationsaktivität
der glatten Muskulatur und/oder Mastzellen-Stabilisationsaktivität und/oder
antiinflammatorischer Aktivität.
Verwendung einer Verbindung nach einem der Ansprüche 13 bis
19 bei der Herstellung eines Medikamentes zum Erreichen von Relaxationsaktivität der glatten
Muskulatur und/oder Mastzellen-Stabilisationsaktivität und/oder
antiinflammatorischer Aktivität.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IE950922 | 1995-12-06 | ||
| IE950922 | 1995-12-06 | ||
| IE960762 | 1996-10-31 | ||
| IE960762 | 1996-10-31 | ||
| PCT/IE1996/000080 WO1997020802A1 (en) | 1995-12-06 | 1996-12-06 | Indane dimer compounds and their pharmaceutical use |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69629387D1 DE69629387D1 (de) | 2003-09-11 |
| DE69629387T2 true DE69629387T2 (de) | 2004-06-24 |
Family
ID=26319864
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69631688T Expired - Fee Related DE69631688T2 (de) | 1995-12-06 | 1996-12-06 | Indanverbindungen mit relaxierender Wirkung auf die glatten Muskeln und/oder mit Mastzellen stabilisierender und/oder entzündungshemmender Wirkung |
| DE69629390T Expired - Fee Related DE69629390T2 (de) | 1995-12-06 | 1996-12-06 | Indandimerverbindungen mit relaxierender wirkung auf die glatten muskeln und/oder mit mastzellen stabilisiernder und/oder entzuendungshemmender wirkung |
| DE69629387T Expired - Lifetime DE69629387T2 (de) | 1995-12-06 | 1996-12-06 | Dimere indan verbindungen und ihre pharmazeutische verwendung |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69631688T Expired - Fee Related DE69631688T2 (de) | 1995-12-06 | 1996-12-06 | Indanverbindungen mit relaxierender Wirkung auf die glatten Muskeln und/oder mit Mastzellen stabilisierender und/oder entzündungshemmender Wirkung |
| DE69629390T Expired - Fee Related DE69629390T2 (de) | 1995-12-06 | 1996-12-06 | Indandimerverbindungen mit relaxierender wirkung auf die glatten muskeln und/oder mit mastzellen stabilisiernder und/oder entzuendungshemmender wirkung |
Country Status (13)
| Country | Link |
|---|---|
| US (3) | US6300376B1 (de) |
| EP (3) | EP0873301B1 (de) |
| JP (3) | JP2000506497A (de) |
| KR (2) | KR19990072007A (de) |
| AT (3) | ATE260241T1 (de) |
| AU (3) | AU1169397A (de) |
| CA (2) | CA2239853C (de) |
| DE (3) | DE69631688T2 (de) |
| DK (1) | DK0865419T3 (de) |
| GB (3) | GB2322858A (de) |
| IE (1) | IE960865A1 (de) |
| IL (3) | IL124756A0 (de) |
| WO (3) | WO1997020805A1 (de) |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2458697A (en) | 1996-04-18 | 1997-11-07 | Alcon Laboratories, Inc. | Calcium channel blockers as human conjunctival mast cell degranulation inhibitors for treating ocular allergic conditions |
| WO1998055439A1 (en) * | 1997-06-05 | 1998-12-10 | Venantius Limited | Indane compounds and their pharmaceutical use |
| JP2001064202A (ja) * | 1999-08-25 | 2001-03-13 | Kissei Pharmaceut Co Ltd | 心不全予防治療剤 |
| DE10142660A1 (de) | 2001-08-31 | 2003-03-20 | Aventis Pharma Gmbh | Verwendung von Derivaten von C2-substituierten Indan-1-ol-Systemen zur Herstellung von Medikamenten zur Prophylaxe oder Behandlung von Obesitas |
| DE10142663B4 (de) * | 2001-08-31 | 2004-08-19 | Aventis Pharma Deutschland Gmbh | C2-Disubstituierte Indan-1-ol-Systeme |
| DE10142667B4 (de) | 2001-08-31 | 2004-06-09 | Aventis Pharma Deutschland Gmbh | C2-substituierte Indan-1-ole und ihre Derivate und ihre Verwendung als Arzneimittel |
| DE10142666A1 (de) | 2001-08-31 | 2003-03-20 | Aventis Pharma Gmbh | Verwendung von C2-substituierten Indan-1-ol-Systemen zur Herstellung von Medikamenten zur Prophylaxe oder Behandlung von Obesitas |
| DE10142668A1 (de) | 2001-08-31 | 2003-03-20 | Aventis Pharma Gmbh | Verwendung von C2-substituierten Indan-1-on-Systemen zur Herstellung von Medikamenten zur Prophylaxe oder Behandlung von Obesitas |
| DE10142722A1 (de) * | 2001-08-31 | 2003-03-27 | Aventis Pharma Gmbh | C2-substituierte Indan-1-one und ihre Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| DE10142661B4 (de) * | 2001-08-31 | 2004-06-09 | Aventis Pharma Deutschland Gmbh | Mehrfach substituierte Indan-1-ol-Systeme und ihre Verwendung als Arzneimittel |
| DE10142665B4 (de) | 2001-08-31 | 2004-05-06 | Aventis Pharma Deutschland Gmbh | C2-Disubstituierte Indan-1-one und ihre Derivate |
| DE10142659A1 (de) | 2001-08-31 | 2003-03-20 | Aventis Pharma Gmbh | Verwendung von mehrfach substituierten Indan-1-ol. Systemen zur Herstellung von Medikamenten zur Prophylaxe oder Behandlung von Obesitas |
| DE10142662B4 (de) | 2001-08-31 | 2004-07-08 | Aventis Pharma Deutschland Gmbh | Derivate von C2-substituierten Indan-1-ol-Systemen und ihre Verwendung als Arzneimittel |
| US10517839B2 (en) * | 2008-06-09 | 2019-12-31 | Cornell University | Mast cell inhibition in diseases of the retina and vitreous |
| US9260376B2 (en) | 2011-07-22 | 2016-02-16 | Venantius Limited | Compounds for use in the treatment of immune related inflammatory disease |
| CN103764608B (zh) | 2011-07-22 | 2016-02-03 | 贝南蒂乌斯有限公司 | 用于治疗炎性肠病的茚衍生物 |
| WO2013014660A1 (en) * | 2011-07-22 | 2013-01-31 | Venantius Limited | Indane dimers for use in the treatment of autoimmune inflammatory disease |
| WO2013174917A1 (en) | 2012-05-24 | 2013-11-28 | Venantius Limited | Compounds for use in the treatment of autoimmune inflammatory disease |
| EP2855416A1 (de) * | 2012-05-24 | 2015-04-08 | Venantius Limited | Indandimere zur verwendung bei der behandlung von autoimmunentzündlichen krankheiten |
| TWI799691B (zh) * | 2019-03-29 | 2023-04-21 | 景凱生物科技股份有限公司 | 具有側鏈烷基與烯基延伸的苯基衍生物及包括有其的藥學組合物 |
| CN111533764B (zh) * | 2020-05-12 | 2023-06-06 | 杭州师范大学 | 一种利用多米诺反应制备硅氧基茚衍生物的方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1205537B (de) * | 1961-08-19 | 1965-11-25 | Thomae Gmbh Dr K | Verfahren zur Herstellung neuer Steroidester |
| US3668258A (en) * | 1969-03-17 | 1972-06-06 | Uniroyal Inc | Sulfur-containing polyaryl polyphenolic compounds and process |
| SE421305B (sv) * | 1973-10-12 | 1981-12-14 | Merck & Co Inc | Forfarande for framstellning av 1-hydroxi-5-indanyloxiettiksyror |
-
1996
- 1996-12-06 AT AT96942552T patent/ATE260241T1/de not_active IP Right Cessation
- 1996-12-06 CA CA002239853A patent/CA2239853C/en not_active Expired - Fee Related
- 1996-12-06 JP JP9521127A patent/JP2000506497A/ja not_active Ceased
- 1996-12-06 GB GB9812215A patent/GB2322858A/en not_active Withdrawn
- 1996-12-06 IL IL12475696A patent/IL124756A0/xx active IP Right Grant
- 1996-12-06 AU AU11693/97A patent/AU1169397A/en not_active Abandoned
- 1996-12-06 DE DE69631688T patent/DE69631688T2/de not_active Expired - Fee Related
- 1996-12-06 AU AU11692/97A patent/AU724648B2/en not_active Ceased
- 1996-12-06 WO PCT/IE1996/000081 patent/WO1997020805A1/en active IP Right Grant
- 1996-12-06 EP EP96942552A patent/EP0873301B1/de not_active Expired - Lifetime
- 1996-12-06 WO PCT/IE1996/000082 patent/WO1997020806A1/en active IP Right Grant
- 1996-12-06 AT AT96942551T patent/ATE246670T1/de not_active IP Right Cessation
- 1996-12-06 GB GB9812213A patent/GB2323088A/en not_active Withdrawn
- 1996-12-06 AU AU11691/97A patent/AU1169197A/en not_active Abandoned
- 1996-12-06 CA CA002239694A patent/CA2239694C/en not_active Expired - Lifetime
- 1996-12-06 WO PCT/IE1996/000080 patent/WO1997020802A1/en active IP Right Grant
- 1996-12-06 DE DE69629390T patent/DE69629390T2/de not_active Expired - Fee Related
- 1996-12-06 GB GB9812216A patent/GB2322859A/en not_active Withdrawn
- 1996-12-06 DE DE69629387T patent/DE69629387T2/de not_active Expired - Lifetime
- 1996-12-06 JP JP9521125A patent/JP2000502328A/ja not_active Ceased
- 1996-12-06 JP JP9521126A patent/JP2000502072A/ja not_active Ceased
- 1996-12-06 AT AT96942550T patent/ATE246668T1/de not_active IP Right Cessation
- 1996-12-06 IE IE960865A patent/IE960865A1/en not_active IP Right Cessation
- 1996-12-06 KR KR1019980704299A patent/KR19990072007A/ko not_active Application Discontinuation
- 1996-12-06 DK DK96942550T patent/DK0865419T3/da active
- 1996-12-06 EP EP96942551A patent/EP0874800B1/de not_active Expired - Lifetime
- 1996-12-06 KR KR1019980704269A patent/KR19990071978A/ko not_active Application Discontinuation
- 1996-12-06 IL IL12479596A patent/IL124795A/xx not_active IP Right Cessation
- 1996-12-06 EP EP96942550A patent/EP0865419B1/de not_active Expired - Lifetime
-
1998
- 1998-06-04 IL IL124756A patent/IL124756A/en not_active IP Right Cessation
- 1998-06-08 US US09/093,060 patent/US6300376B1/en not_active Expired - Lifetime
- 1998-06-08 US US09/092,903 patent/US6423752B1/en not_active Expired - Fee Related
- 1998-06-08 US US09/092,902 patent/US6297399B1/en not_active Expired - Fee Related
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69629387T2 (de) | Dimere indan verbindungen und ihre pharmazeutische verwendung | |
| AU599029B2 (en) | Ascorbic acid derivatives, production and use thereof | |
| AT390732B (de) | Pharmazeutika enthaltend pyrazole | |
| US4578498A (en) | Phenylethylene derivatives and their use as drugs | |
| EP1655280A1 (de) | Neuartige Dicarbonsäurederivate mit pharmazeutischen Eigenschaften | |
| DE3535167A1 (de) | Neue sulfonyl-phenyl(alkyl)amine, verfahren zu ihrer herstellung sowie arzneimittel | |
| EP0600949B1 (de) | Neue 3,5-di-tert.butyl-4-hydroxyphenyl-derivate, verfahren zu ihrer herstellung und arzneimittel | |
| EP0084667B1 (de) | Phenylethylen-Derivate, ihre Herstellung und Verwendung als Arzneimittel | |
| CH494730A (de) | Verfahren zur Herstellung von Dibenzocyclopentenen | |
| CH633250A5 (de) | Verfahren zur herstellung von neuen polyprenylderivaten. | |
| DE60121809T2 (de) | Materialien und verfahren zur herstellung von stilbenen | |
| EP0471259B1 (de) | Phenylsulfonamid-substituierte Pyridinalken- und -aminooxyalkancarbonsäure-derivate | |
| EP0298919A2 (de) | 4-Amino-substituierte 1,2-Dihydroxynaphthalin-Derivate | |
| DE2929760C2 (de) | ||
| DE60112917T2 (de) | Neue polyzyklische indanylimidazole mit alpha2 adrenergik aktivität | |
| DE2332707A1 (de) | Tricyclische tetrahydronaphthalinderivate, ihre salze, verfahren zu ihrer herstellung und diese verbindungen enthaltende arzneimittel | |
| DE69633289T2 (de) | 4-acylamino(halogen)alkyl-chinolin derivate, deren herstellung und deren verwendung als melatonin-agonisten | |
| DE2164662A1 (de) | Indanderivate und Verfahren zu ihrer Herstellung | |
| EP0433806B1 (de) | Isocyanatoalkylsulfonate und ein Verfahren zu deren Herstellung | |
| EP0891322B1 (de) | Dimethyl-substituierte cyclohexandienderivate | |
| DE3617938A1 (de) | Neue isochinolin-derivate, verfahren zu ihrer herstellung und pharmazeutische zusammensetzungen | |
| DE10047486A1 (de) | Phenoxyphenyl Alkansulfonate | |
| AT360992B (de) | Verfahren zur herstellung neuer phenylazacyclo- alkane und von deren salzen und optisch aktiven verbindungen | |
| DE2264663C3 (de) | Ungesättigte Sulfoxyde und Verfahren zu ihrer Herstellung | |
| EP0413692B1 (de) | Verwendung von hexatrienderivaten zur herstellung von mitteln gegen akne, psoriasis und lichtschäden der haut |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition |