Verfahren zur Herstellung von monoglycosidierten Flavonoiden Process for the preparation of monoglycosidated flavonoids
Die vorliegende Erfindung betrifft ein Verfahren zur Herstellung von monoglycosidierten Flavonoiden durch enzymatische Hydrolyse von Rutinosiden. Dabei wird der Rhamno- serest der Rutinoside enzymatisch abgespalten.The present invention relates to a process for the preparation of monoglycosidated flavonoids by enzymatic hydrolysis of rutinosides. The rhamno residue of the rutinosides is cleaved off enzymatically.
Im Rahmen der vorliegenden Erfindung werden als Rutinoside solche Verbindungen bezeichnet, die einen zuckerfreien Bestandteil enthalten, an welchen ein Rest der Formel (I)
In the context of the present invention, rutinosides are compounds which contain a sugar-free component in which a radical of the formula (I)
über eine glycosidische Bindung gebunden ist. Beispielsweise handelt es sich bei den Rutinosiden um Flavonoide mit der in Formel (I) dargestellten bisgylcosidischen Einheit. Rhamnose und/oder die entsprechenden Glucopyranoside können aus den Rutinosiden gewonnen werden. Die Glucopyranoside leiten sich von den Rutinosiden dadurch ab, dass sie anstelle des Rests der Formel (I) einen Rest der Formel (I*)
is bound via a glycosidic bond. For example, the rutinosides are flavonoids with the bisgylcosidic unit shown in formula (I). Rhamnose and / or the corresponding glucopyranosides can be obtained from the rutinosides. The glucopyranosides are derived from the rutinosides in that instead of the residue of the formula (I) they contain a residue of the formula (I *)
enthalten, der an den zuckerfreien Bestandteil gebunden ist. Beispielsweise können aus Rutin sowohl Rhamnose als auch Isoquercetin gewonnen werden.
Rhamnose ist ein Monosaccharid, welches in der Natur weit verbreitet, allerdings zumeist nur in geringen Mengen vorkommt. Eine wichtige Quelle für Rhamnose sind z.B. die glycosidischen Reste natürlicher Flavonoide, wie Rutin, aus welchen die Rhamnose durch Glycosidspaltung gewonnen werden kann. Rhamnose spielt beispielsweise eine wichtige Rolle als Ausgangsstoff für die Darstellung von künstlichen Aromastoffen, wie Furaneol.included, which is bound to the sugar-free ingredient. For example, both rhamnose and isoquercetin can be obtained from rutin. Rhamnose is a monosaccharide, which is widespread in nature, but mostly occurs only in small amounts. An important source for rhamnose are, for example, the glycosidic residues of natural flavonoids, such as rutin, from which the rhamnose can be obtained by glycoside cleavage. Rhamnose, for example, plays an important role as a raw material for the representation of artificial flavors, such as furaneol.
Isoquercetin ist ein monoglycosidiertes Flavonoid der folgenden Strukturformel (II)
Isoquercetin is a monoglycosidated flavonoid of the following structural formula (II)
Als Flavonoide (lat. flavu = gelb), die in Pflanzen weit verbreitete Farbstoffe sind, werden z.B. Glycoside von Flavonen, denen das Grundgerüst des Flavons (2-Phenyl-4H-1- benzopyran-4-on) gemeinsam ist, bezeichnet.As flavonoids (lat. Flavu = yellow), which are widely used dyes in plants, e.g. Glycosides of flavones which share the basic structure of flavone (2-phenyl-4H-1-benzopyran-4-one).
Der zuckerfreie Bestandteil der Flavonoide ist das sogenannte Agiykon. Isoquercetin ist beispielsweise ein Glycosid des Aglykons Quercetin (2-(3,4-Dihydrophenyl)-3,5,7- trihydroxy-4H-1-benzopyran-4-on), welches sich von Flavon durch das Vorhandensein von fünf Hydroxylgruppen unterscheidet. Im Isoquercetin ist der Kohlenhydratrest Glu- cose an die Hydroxylgruppe in Position 3 des Quercetins gebunden. Isoquercetin wird z.B. als Quercetin-3-O-ß-D-glucopyranosid oder 2-(3,4-Dihydroxyphenyl)-3-(ß-D- glucopyranosyloxy)-5,7-dihydroxy-4H-1-benzopyran-4-on bezeichnet. Es ist aber beispielsweise auch unter dem Namen Hirsutrin bekannt.
Flavonoide bzw. Flavonoidgemische werden beispielsweise in der Lebensmittel- und Kosmetikindustrie verwendet und gewinnen dort zunehmend an Bedeutung. Besonders monoglycosidierte Flavonoide, wie z.B. Isoquercetin, zeichnen sich durch eine gute Aufnahmefähigkeit im menschlichen Körper aus.The sugar-free component of the flavonoids is the so-called agiykon. For example, isoquercetin is a glycoside of the aglycon quercetin (2- (3,4-dihydrophenyl) -3,5,7-trihydroxy-4H-1-benzopyran-4-one), which differs from flavone in that it contains five hydroxyl groups. In isoquercetin, the carbohydrate residue glucose is bound to the hydroxyl group in position 3 of quercetin. For example, isoquercetin is used as quercetin-3-O-ß-D-glucopyranoside or 2- (3,4-dihydroxyphenyl) -3- (ß-D-glucopyranosyloxy) -5,7-dihydroxy-4H-1-benzopyran-4- called on. However, it is also known, for example, under the name hirsutrin. Flavonoids or flavonoid mixtures are used, for example, in the food and cosmetics industry and are becoming increasingly important there. Especially monoglycosidated flavonoids, such as isoquercetin, are characterized by their good absorption capacity in the human body.
Ein Beispiel für ein natürlich vorkommendes Flavonoid mit bisglycosidischer Einheit ist Rutin, welches folgende Strukturformel (III) besitzt:
An example of a naturally occurring flavonoid with a bisglycosidic unit is rutin, which has the following structural formula (III):
Rutin ist wie Isoquercetin ebenfalls ein Glycosid des Aglykons Quercetin, wobei der Kohlenhydratrest Rutinose an die Hydroxylgruppe in Position 3 des Quercetins gebunden ist. Der Kohlenhydratrest im Rutin besteht aus einer in 1- und 6-Stellung verknüpften Glucose- und einer terminal gebundenen Rhamnose- bzw. 6-Deoxymannose- Einheit. Rutin wird z.B. als Quercetin-3-O-ß-D-rutinosid oder 2-(3,4-Dihydroxyphenyl)-3- { [6-O-(6-deoxy-α-mannopyranosol)-ß-D-glucopyranosyl]oxy !>-5,7-dihydroxy-4H-1 - benzopyran-4-on bezeichnet. Es ist jedoch beispielsweise auch unter den Namen Sophorin, Birutan, Rutabion, Taurutin, Phytomelin, Melin oder Rutosid bekannt.Like isoquercetin, rutin is also a glycoside of the aglycone quercetin, the carbohydrate residue rutinose being bound to the hydroxyl group in position 3 of the quercetin. The carbohydrate residue in rutin consists of a glucose unit linked in the 1- and 6-position and a terminally bound rhamnose or 6-deoxymannose unit. Rutin is used, for example, as quercetin-3-O-ß-D-rutinoside or 2- (3,4-dihydroxyphenyl) -3- {[6-O- (6-deoxy-α-mannopyranosol) -ß-D-glucopyranosyl] oxy! > -5,7-dihydroxy-4H-1-benzopyran-4-one. However, it is also known, for example, under the names sophorin, birutan, rutabion, taurutin, phytomelin, melin or rutoside.
Rutin bildet mit drei Molekülen Kristallwasser blassgelbe bis grünliche Nadeln. Wasserfreies Rutin hat die Eigenschaft einer schwachen Säure, wird bei 125°C braun und zersetzt sich bei 214-215°C. Rutin, das in vielen Pflanzenarten - häufig als Begleiter
des Vitamins C - vorkommt, z.B. in Zitrusarten, in gelben Stiefmütterchen, Forsythien-, Akazienarten, verschiedene Solanum- und Nicotiana-Arten, Kapern, Lindenblüten, Johanniskraut, Tee usw., wurde 1842 aus der Gartenraute (Ruta graveolens) isoliert. Rutin kann auch den Blättern des Buchweizens und der ostasiatischen Färberdroge Wei-Fa (Sophora japonica, Fabaceae), die 13-27% Rutin enthält, gewonnen werden.With three molecules of water of crystallization, rutin forms pale yellow to greenish needles. Anhydrous rutin has the property of a weak acid, turns brown at 125 ° C and decomposes at 214-215 ° C. Rutin, which is found in many plant species - often as a companion of vitamin C - occurs, for example, in citrus, in yellow pansies, forsythia, acacia, various Solanum and Nicotiana species, capers, linden flowers, St. John's wort, tea, etc., was isolated in 1842 from the garden rhombus (Ruta graveolens). Rutin can also be obtained from the leaves of buckwheat and the East Asian coloring drug Wei-Fa (Sophora japonica, Fabaceae), which contains 13-27% rutin.
Es ist wünschenswert, sowohl Rhamnose als auch monoglycosidierte Flavonoid aus natürlichen Rohstoffen, beispielsweise aus Flavonoiden mit einer bisglycosidischen Einheit, herzustellen. In diesem Zusammenhang ist z.B. die Spaltung von Rutinosiden zu Rhamnose und den entsprechenden Glucopyranosiden interessant.It is desirable to produce both rhamnose and monoglycosidated flavonoid from natural raw materials, for example from flavonoids with a bisglycosidic unit. In this context, e.g. the splitting of rutinosides into rhamnose and the corresponding glucopyranosides is interesting.
Enzymatisch katalysierte Darstellungen von Rhamnose sind in der Literatur beschrieben. Beispielsweise sind in der EP-A-0317033 ein Verfahren zur Herstellung von L-Rhamnose beschrieben, wobei die rhamnosidische Bindung von Glycosiden, die Rhamnose in terminaler Position gebunden halten, durch enzymatische Hydrolyse erreicht wird. Bei dieser Spaltung wird das Substrat üblicherweise als Suspension in einem wässrigen Medium durchgeführt. Jedoch sind diese Reaktionen zumeist wenig selektiv. Beispielsweise entsteht aufgrund der bisglycosidischen Struktur des Kohlenhydratrestes im Rutin oft eine Mischung der beiden Monosaccharide Glucose und Rhamnose. Zudem entstehen zumeist hohe Anteile am Agiykon Quercetin sowie weitere unerwünschte Nebenprodukte.Enzymatically catalyzed representations of rhamnose are described in the literature. For example, EP-A-0317033 describes a process for the preparation of L-rhamnose, the rhamnosidic binding of glycosides which keep rhamnose bound in the terminal position is achieved by enzymatic hydrolysis. In this cleavage, the substrate is usually carried out as a suspension in an aqueous medium. However, these reactions are mostly not very selective. For example, due to the bisglycosidic structure of the carbohydrate residue in rutin, a mixture of the two monosaccharides glucose and rhamnose is often formed. In addition, there is usually a high proportion of Agiykon quercetin and other undesirable by-products.
Weiterhin werden enzymatisch katalysierte Spaltungen von Rutin auch in der JP-A-01213293 beschrieben. Allerdings verlaufen derartige in wässrigen Medien durchgeführte Reaktionen zumeist ebenfalls wenig selektiv.Furthermore, enzymatically catalyzed cleavages of rutin are also described in JP-A-01213293. However, reactions of this type carried out in aqueous media are usually also not very selective.
Diese vorstehend beschriebenen Verfahren verwenden das Enzym in Lösung, d.h. na- tiv. Dabei wird das Enzym direkt der Reaktionslösung zugegeben. Obwohl diese Verfahren im Labormaßstab durchführbar sind, sind sie für eine technische Anwendung nicht praktikabel, da das Enzym aus der Reaktionslösung zur erneuten Verwendung nicht zurückgewonnen werden kann. Der einmalige Gebrauch dieser teuren Enzyme ist jedoch bei einer technischen Anwendung nicht wirtschaftlich.
Es ist bekannt, dass Enzyme technisch einsetzbar sind, indem sie an einen Träger gebunden werden. Dieses Verfahren wird als „Immobilisieren" bezeichnet. Unter dem Begriff „immobilisierte Enzyme" werden durch die European Federation of Biotechnology (1983) alle Enzyme zusammengefasst, "... die sich in einem Zustand befinden, der ihre Wiederverwendung erlaubt" (Helmut Uhlig, Technische Enzyme und ihre Anwendung, Carl Hanser Verlag, München Wien 1991 , S. 198). Trotz dieses Vorteils ist eine Immobilisierung jedoch nicht für alle Enzymprozesse geeignet und hat sich bisher auch nur begrenzt durchgesetzt. Insbesondere finden nur zwei immobilisierte Enzyme großtechnisch Anwendung: Die Glucoseisomerisierung mit immobilisierter Glucoseisomerase und die Peniciliin-G-Spaltung mit immobilisierter Penicillinamidase. Oft können immobilisierte Enzymverfahren nicht gegenüber den freien Enzymen oder chemischen Verfahren bestehen. Oft sind auch die Enzyme oder die Reaktionsbedingungen für eine Immobilisierung nicht geeignet. Es gibt also keine Universalmethode für das Immobilisieren, und jedes Enzym muss individuell betrachtet werden.These methods described above use the enzyme in solution, ie native. The enzyme is added directly to the reaction solution. Although these processes can be carried out on a laboratory scale, they are not practical for industrial use because the enzyme cannot be recovered from the reaction solution for reuse. However, the one-time use of these expensive enzymes is not economical in a technical application. It is known that enzymes can be used industrially by binding them to a carrier. This process is referred to as "immobilization". The term "immobilized enzymes" is used by the European Federation of Biotechnology (1983) to summarize all enzymes "... that are in a state that permits their reuse" (Helmut Uhlig, Technical enzymes and their application, Carl Hanser Verlag, Munich Vienna 1991, p. 198). Despite this advantage, immobilization is, however, not suitable for all enzyme processes and has so far only been used to a limited extent. In particular, only two immobilized enzymes are used on an industrial scale: glucose isomerization with immobilized glucose isomerase and peniciliin G cleavage with immobilized penicillin amidase. Often immobilized enzyme processes cannot compete with free enzymes or chemical processes. The enzymes or the reaction conditions are often also unsuitable for immobilization. So there is no universal method for immobilization, and each enzyme must be viewed individually.
Beispielsweise kommt es in wässrigen Systemen, wie sie für die Funktionstüchtigkeit von Enzymen wichtig sind, zu Löslichkeitsproblemen bei Rutinosiden, die bei der enzy- matischen Hydrolyse als Substrat dienen. Diese Reaktion wird deshalb vorzugsweise mit einer übersättigten Substratlösung, d.h. als Rutinosidsuspension, durchgeführt. Bei einer übersättigten Lösung, in der das Substrat als Feststoff vorliegt, ist jedoch eine Anwendung der Immobilisierungstechniken unmöglich. Es mangelt dabei an der Selektivität zwischen Rohstoff und Produktpartikeln und feststoffgebundenem Enzym.For example, in aqueous systems, which are important for the functionality of enzymes, there are solubility problems with rutinosides, which serve as a substrate in the enzymatic hydrolysis. This reaction is therefore preferably carried out with a supersaturated substrate solution, i.e. as a rutinoside suspension. In the case of a supersaturated solution in which the substrate is present as a solid, however, immobilization techniques cannot be used. There is a lack of selectivity between raw material and product particles and solid-bound enzyme.
Es ist deshalb die Aufgabe der vorliegenden Erfindung, ein Verfahren zur Herstellung von monoglycosidierten Enzymen zur Verfügung zu stellen, das im technischen Maßstab angewendet werden kann, wobei hohe Enzymkosten vermieden werden und gleichzeitig ein hoher Automatisierungsgrad verbunden mit einer hohen Raum/Zeit- Ausbeute und einer hohen Produktivität und Selektivität erreicht wird. Diese Aufgabe wird gelöst durch ein Verfahren zur Herstellung von monoglycosidierten Flavonoiden durch enzymatische Hydrolyse von Rutionosiden, worin für die enzymatische Hydrolyse ein Enzym, das auf einem Träger immobilisiert ist, verwendet wird.
Überraschend wurde gefunden, dass trotz einer geringen Löslichkeit von Rutinosiden eine enzymatische Hydrolyse mit einem immobilisierten Enzym möglich ist. Durch die Immobilisierung kann das Verfahren kontinuierlich oder diskontinuierlich und mit einer hohen Effektivität im Vergleich zu der Reaktion mit dem nativen Enzym durchgeführt werden. Das erfindungsgemäße Verfahren zeichnet sich insbesondere dadurch aus, dass eine hohe Automatisierung des gesamten Verfahrens einschließlich der Rückführung des Lösungsmittels sowie der Überwachung der Enzymaktivität möglich ist.It is therefore the object of the present invention to provide a process for the production of monoglycosidated enzymes which can be used on an industrial scale, high enzyme costs being avoided and, at the same time, a high degree of automation combined with a high space / time yield and high productivity and selectivity is achieved. This object is achieved by a process for the production of monoglycosidated flavonoids by enzymatic hydrolysis of rutionosides, in which an enzyme which is immobilized on a support is used for the enzymatic hydrolysis. Surprisingly, it was found that, despite the low solubility of rutinosides, enzymatic hydrolysis with an immobilized enzyme is possible. Due to the immobilization, the process can be carried out continuously or discontinuously and with a high effectiveness in comparison with the reaction with the native enzyme. The process according to the invention is characterized in particular by the fact that high automation of the entire process, including the recycling of the solvent and the monitoring of the enzyme activity, is possible.
In Abbildung 1 wird als Beispiel des erfindungsgemäßen Verfahrens die kontinuierliche Herstellung von Isoquercetin aus Rutin dargestellt.Figure 1 shows the continuous production of isoquercetin from rutin as an example of the process according to the invention.
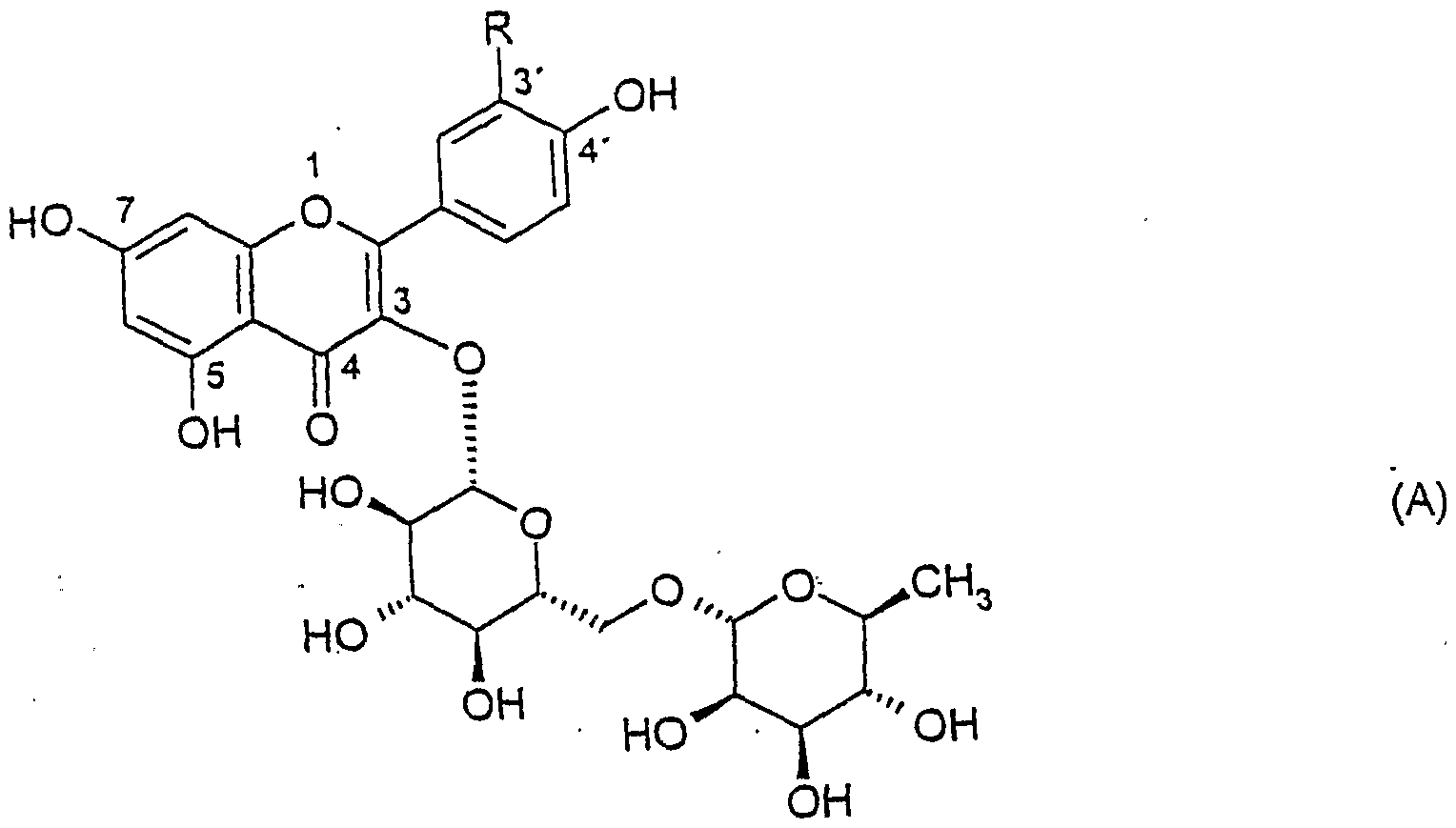
Geeignete Rutinoside für das erfindungsgemäße Verfahren sind jene, die als zuckerfreien Bestandteil oder Agiykon einen 2-Phenyl-4H-1-benzopyran-4-on-Grundkörper enthalten, der in der Position 3 einen Rest der Formel (I) trägt und dessen Phenylgrup- pen, abgesehen von der Position 3, auch ein- oder mehrfach durch -OH oder -0(CH2)n-H, wobei n 1 bis 8 bedeutet, substituiert sein können, n bedeutet vorzugsweise 1.Suitable rutinosides for the process according to the invention are those which contain a 2-phenyl-4H-1-benzopyran-4-one base body as a sugar-free constituent or agiycon, which bears a radical of the formula (I) in position 3 and whose phenyl group pen, apart from position 3, can also be substituted one or more times by -OH or -0 (CH 2 ) n -H, where n is 1 to 8, n is preferably 1.
Die Substitution des 2-Phenyl-4H-1 -benzopyran-on-Grundkörpers durch -OH und/oder -0(CH2)n-H tritt bevorzugt in den Positionen 5, 7, 3' und/oder 4' auf.The substitution of the 2-phenyl-4H-1-benzopyran-one base body by -OH and / or -0 (CH 2 ) n -H preferably occurs in positions 5, 7, 3 'and / or 4'.
Besonders bevorzugt werden Rutinoside verwendet, die durch die Formel (A) dargestellt werden:
Rutinosides, which are represented by the formula (A), are particularly preferably used:
worin R H, OH oder OCH3 darstellt.wherein RH, OH or OCH represents 3 .
Die Verbindung, worin R H darstellt, wird als Kämpferolrutinosid bezeichnet; das Ruti- nosid, worin R OCH3 darstellt, ist als Isorhamnetinrutinosid bekannt. Die Verbindung, worin R OH bedeutet, wird als Rutin bezeichnet. Demzufolge können gemäß dem erfindungsgemäßen Verfahren aus Kämpferolrutinosid Rhamnose und Kämpferolglucosid, aus Rutin Rhamnose und Isoquercetin und aus Isorhamnetinrutinosid Rhamnose und Isorhamnetinglycosid erhalten werden.The compound in which RH represents is referred to as fighter olrutinoside; the rutinoside, in which R represents OCH 3 , is known as isorhamnetin rutinoside. The compound in which R is OH is called rutin. Accordingly, rhamnose and fighter olglucoside can be obtained from fighter olrutinoside, rhamnose and isoquercetin from rutin rutinoside and rhamnose and isorhamnetin glycoside from isorhamnetin rutinoside.
Besonders bevorzugt wird das Rutinosid Rutin verwendet.The rutinoside rutin is particularly preferably used.
Als Ausgangsmaterial für das erfindungsgemäße Verfahren können die Rutinoside in reiner Form oder auch als Gemische von Rutinosiden eingesetzt werden. Ebenfalls können die Rutinoside mit anderen Flavonoiden oder mit Rückständen aus der Rutino- sidproduktion verunreinigt sein, ohne dass die Reaktion negativ beeinflusst wird.The rutinosides can be used in pure form or as mixtures of rutinosides as starting material for the process according to the invention. The rutinosides can also be contaminated with other flavonoids or with residues from rutinoside production without the reaction being adversely affected.
Als Enzyme für die enzymatische Hydrolyse der Rutinoside können übliche Hydrolasen verwendet werden, die in der Lage sind, den Rhamnoserest der Rutinoside abzuspalten. Vorzugsweise werden Hydrolasen, die aus dem Stamm Penicillium decumbens
gewonnen werden, verwendet. Besonders bevorzugt werden als Enzyme α-L- Rhamnosidasen verwendet, da diese eine hohe Selektivität für die Hydrolyse des Rhamnoserestes besitzen. Geeignete α-L-Rhamnosidasen sind beispielsweise Hesperidinase, Naringinase, sowie jene, die in Kurosawa et al. (1973), J. Biochem., Bd.. 73: 31-37 beschrieben werden. Es ist besonders bevorzugt, das Enzym Hesperidinase zu verwenden.Usual hydrolases which are able to split off the rhamnose residue of the rutinosides can be used as enzymes for the enzymatic hydrolysis of the rutinosides. Hydrolases originating from the strain Penicillium decumbens are preferred won, used. Α-L-Rhamnosidases are particularly preferably used as enzymes, since they have a high selectivity for the hydrolysis of the rhamnose residue. Suitable α-L-rhamnosidases are, for example, hesperidinase, naringinase, and those described in Kurosawa et al. (1973), J. Biochem., Vol. 73: 31-37. It is particularly preferred to use the enzyme hesperidinase.
Sowohl die Rutinoside als auch die Enzyme für das erfindungsgemäße Verfahren können als Handelsprodukte erworben werden. Ebenfalls ist es möglich, die Ausgangsstoffe und Enzyme nach allgemein bekannten Methoden zu gewinnen oder herzustellen.Both the rutinosides and the enzymes for the process according to the invention can be purchased as commercial products. It is also possible to obtain or produce the starting materials and enzymes by generally known methods.
Das Enzym wird auf einem geeigneten Träger immobilisiert. Dabei können übliche Träger, wie Kieselgel, beispielsweise handelsübliche sphärische oder handelsübliche gebrochene Kieselgele, z.B. Lichrosorb®, Lichroprep®, Lichrospher® und Trisoperl®, und handelsübliche polymere Träger, z.B. Eupergit®, Fractogel®, insbesondere Fractogel epoxy®,und Fractoprep®, zum Einsatz kommen. Kieselgel gilt als bevorzugtes Trägermaterial.The enzyme is immobilized on a suitable carrier. Typical carriers, such as silica gel, for example commercially available spherical or commercially available broken silica gels, for example Lichrosorb ® , Lichroprep ® , Lichrospher ® and Trisoperl ® , and commercially available polymeric carriers, for example Eupergit ® , Fractogel ® , in particular Fractogel epoxy ® , and Fractoprep ® , are used. Silica gel is the preferred carrier material.
Alternativ können auch magnetische Partikel als Träger verwendet werden. Vorzugsweise handelt es sich dabei um Trägermaterialien mit einem magnetischen Kern. Dieser Kern ist üblicherweise von einem anorganischen Oxid umhüllt. Das anorganische Oxid ist vorzugsweise Kieselgel. Beispiele derartiger magnetischer Träger umfassen Magne- Sil™ (Promega Corp., Madison, Wisconsin, US), MagPrep™ (Merck) und AGOWAmag™ (AGOWA GmbH, Berlin, DE). Als magnetische Träger können aber auch magnetische Glaspartikel (z.B. MPG (CPG Inc., Lincoln Park, New Jersey, US)), sowie magnetithalti- ge Pigmente (z.B. Microna Matte, Mica Black, Colorona Blackstar (alle Merck)) eingesetzt werden. Besonders gut eignen sich unporöse magnetische Partikel (wie MagPrep), da es bei diesen nicht zu einer Porenverblockung kommen kann, wodurch sich die Enzymaktivität drastisch verschlechtern würde.Alternatively, magnetic particles can also be used as carriers. These are preferably carrier materials with a magnetic core. This core is usually coated with an inorganic oxide. The inorganic oxide is preferably silica gel. Examples of such magnetic carriers include MagneSil ™ (Promega Corp., Madison, Wisconsin, US), MagPrep ™ (Merck) and AGOWAmag ™ (AGOWA GmbH, Berlin, DE). Magnetic glass particles (e.g. MPG (CPG Inc., Lincoln Park, New Jersey, US)) and magnetite-containing pigments (e.g. Microna Matte, Mica Black, Colorona Blackstar (all Merck)) can also be used as magnetic carriers. Non-porous magnetic particles (such as MagPrep) are particularly well suited because they cannot block pores, which would drastically impair the enzyme activity.
Der Enzymträger besitzt üblicherweise die folgenden Eigenschaften: Die Teilchengröße des Trägers beträgt vorzugsweise 0,005 bis 1 mm, noch bevorzugter 0,01 bis 0,5 mm.
Der Porendurchmesser liegt üblicherweise im Bereich von 10 bis 4000 nm, wobei ein Porendurchmesser von 30 bis 100 nm besonders bevorzugt ist. Durch eine ausreichend große Porengröße kann sichergestellt werden, dass das Enzym ohne Aktivitätsverlust auf dem Träger Platz findet. Die Partikeloberfläche beträgt vorteilhafterweise 40 bis 100 m2/g, und das Porenvolumen wird vorzugsweise aus einem Bereich von 0,5 bis 3 ml/g ausgewählt. In manchen Fällen kann auch ein sehr großer Porendurchmesser von 2 bis 20 μm geeignet sein.The enzyme carrier usually has the following properties: The particle size of the carrier is preferably 0.005 to 1 mm, more preferably 0.01 to 0.5 mm. The pore diameter is usually in the range from 10 to 4000 nm, a pore diameter from 30 to 100 nm being particularly preferred. A sufficiently large pore size can ensure that the enzyme fits on the support without loss of activity. The particle surface is advantageously 40 to 100 m 2 / g, and the pore volume is preferably selected from a range of 0.5 to 3 ml / g. In some cases, a very large pore diameter of 2 to 20 μm can also be suitable.
Das Enzym kann kovalent oder adsorptiv gekuppelt werden. Generell ist eine kovalente Kupplung vorzuziehen. Beispiele einer kovalenten Kupplung beinhalten eine Epoxidie- rung, eine Carbodiimidmethode, Silanisierung, eine Bromcyanmethode, eine Glutar- dialdehydvemetzung oder eine Dicresylchloridmethode (siehe Biotransformations and Enzyme Reactions, Bommarius, A.S., Biotechnology (2. Aufl.), Bd.3, S. 427-465, zusammengestellt von Stephanopoulos, G., VCH Weinheim, Deutschland 1993, Walt, D.R. et al., trends in analytical chemistry, Bd. 13, Nr. 10, 1994, N.H. Park, H.N. Chang; J. Ferment. Technol., Bd. 57 (4), 310-316, 1979, M. Puri et al.; Enz. Microb. Technol., 18, 281-285, 1996 und H-Y. Tsen; J. Ferment. Technol., 62 (3), 263-267, 1984). Zur Durchführung dieser Verfahren ist es notwendig, dass der Träger mit entsprechenden funktionellen Gruppen oberflächenmodifiziert wird. Die funktioneile Gruppe kann am Träger entweder durch Copolymerisation mit funktionellen Monomeren oder durch Polymeranalogumsetzung vollzogen werden. Besonders bevorzugt ist eine Oberflächenmodifikation mit Aminoresten, Aldehydgruppen, Epoxygruppen, oder eine Diolmodifizie- rung. An diese Gruppen können dann die Enzyme kovalent gebunden werden.The enzyme can be coupled covalently or adsorptively. Generally, a covalent coupling is preferable. Examples of a covalent coupling include epoxidation, a carbodiimide method, silanization, a cyanogen bromide method, a glutaraldehyde crosslinking or a dicresyl chloride method (see Biotransformations and Enzyme Reactions, Bommarius, AS, Biotechnology (2nd ed.), Vol. 3, p. 427-465, compiled by Stephanopoulos, G., VCH Weinheim, Germany 1993, Walt, DR et al., Trends in analytical chemistry, Vol. 13, No. 10, 1994, NH Park, HN Chang; J. Ferment. Technol ., Vol. 57 (4), 310-316, 1979, M. Puri et al .; Enz. Microb. Technol., 18, 281-285, 1996 and HY. Tsen; J. Ferment. Technol., 62 ( 3), 263-267, 1984). To carry out these processes, it is necessary for the support to be surface-modified with appropriate functional groups. The functional group can be carried out on the support either by copolymerization with functional monomers or by polymer analog conversion. A surface modification with amino residues, aldehyde groups, epoxy groups or a diol modification is particularly preferred. The enzymes can then be covalently bound to these groups.
Die enzymatische Hydrolyse findet in einem geeigneten Reaktor statt. Für eine kontinuierliche Durchführung des erfindungsgemäßen Verfahrens eignet sich insbesondere eine handelsübliche Säule. Im kleinen Maßstab kann beispielsweise eine Säule, wie sie für die präparative HPLC verwendet wird, eingesetzt werden. Der Reaktor, insbesondere die Säule, sollte einen hohen hydraulischen Wirkungsgrad aufweisen. Dies kann durch die Zahl der theoretischen Böden quantifiziert werden. Deshalb ist es von Vorteil, dass es zu einem intensiven Kontakt der Rohstofflösung mit der Oberfläche des Immo- bilisats kommt, um die wirksame Nutzung des Enzyms zu gewährleisten und eine hohe Produktivität zu erreichen. Die erwähnte präparative HPLC-Säule erfüllt diese Anforde-
rungen und ist ebenfalls mit der entsprechenden Technik mit Peripherie (Pumpen, Ventile, Steuerung) ausgestattet. Es ist ebenfalls von Vorteil, dass hierfür bereits Detekti- onstechniken, wie UV- oder Rl-Detektionstechniken entwickelt wurden, so dass, wenn erwünscht, die Messung und Regelung des Umsatzgrades der Reaktion automatisiert durchgeführt werden kann.The enzymatic hydrolysis takes place in a suitable reactor. A commercially available column is particularly suitable for a continuous implementation of the method according to the invention. On a small scale, for example, a column such as that used for preparative HPLC can be used. The reactor, especially the column, should have a high hydraulic efficiency. This can be quantified by the number of theoretical floors. It is therefore advantageous that the raw material solution comes into intensive contact with the surface of the property in order to ensure effective use of the enzyme and to achieve high productivity. The preparative HPLC column mentioned fulfills these requirements. and is also equipped with the appropriate technology with peripherals (pumps, valves, control). It is also advantageous that detection techniques such as UV or RI detection techniques have already been developed for this, so that, if desired, the measurement and regulation of the degree of conversion of the reaction can be carried out automatically.
Werden bei der kontinuierlichen Fahrweise magnetische Trägermaterialien eingesetzt, werden üblicherweise Rohrreaktoren mit einer Vorrichtung eingesetzt, die die magnetischen Partikel stabil in Schwebe halten, z.B. elektromagnetische Spulen, die im Strömungsrohr ein weitgehend homogenes Magnetfeld erzeugen, dessen Feldlinien parallel zur Strömungsrichtung verlaufen (Helmholtz-Magnetfeld). In solchen magnetisch stabilisierten Wirbelschicht- oder Fließbett-Reaktoren {Magnetically stabilized fluidized bed (MSFB)) können wesentlich höhere Strömungsgeschwindigkeiten erreicht werden als in konventionellen Wirbelschichten oder Fließbettkolonnen, die aber auch für diese Zwecke geeignet sind. Diese Technologie kann auch vorteilhaft für Katalysereaktionen in viskosen Reaktionsmedien eingesetzt werden.If magnetic carrier materials are used in the continuous mode of operation, tubular reactors are usually used with a device which keeps the magnetic particles in suspension, e.g. electromagnetic coils that generate a largely homogeneous magnetic field in the flow tube, the field lines of which run parallel to the direction of flow (Helmholtz magnetic field). In such magnetically stabilized fluidized bed or fluidized bed reactors (Magnetically Stabilized Fluidized Bed (MSFB)), significantly higher flow rates can be achieved than in conventional fluidized beds or fluidized bed columns, but these are also suitable for these purposes. This technology can also be used advantageously for catalytic reactions in viscous reaction media.
Für ein diskontinuierliche Durchführung des Verfahrens eignet sich ein üblicher Behälter, der vorzugsweise mit einer Rührvorrichtung ausgestattet ist. So kann im kleinen Maßstab ein Rundkolben mit Rührer und im großen Maßstab ein Rührkessel eingesetzt werden.A conventional container, which is preferably equipped with a stirring device, is suitable for carrying out the process discontinuously. A round bottom flask with a stirrer can be used on a small scale and a stirred kettle on a large scale.
Das Immobilisat wird vor der Reaktion auf übliche Weise in den Reaktor gepackt.The immobilizate is packed into the reactor in the usual way before the reaction.
Das umzusetzende Rutinosid wird in den Reaktor, z.B eine Säule, wie eine Festbettsäule, üblicherweise in Form einer Lösung oder Suspension gespeist. Handelt es sich bei dem Reaktor um einen Festbettreaktor sollte die Rutinosidlösung vollkommen feststofffrei sein. Es ist dabei von Vorteil, das Rutinosid mit dem Lösungsmittel in einem Tank, vorzugsweise unter Rühren und/oder Erwärmen, vorzulösen, um eine optimale Löslichkeit zu erreichen. Wenn notwendig, kann außerdem eine Vorfiltration der Lösung durchgeführt werden, um mögliche Feststoffe zu entfernen. Das Lösungsmittel ist vorzugsweise ein wässriges System, um die Enzymaktivität zu gewährleisten und mögliche Denaturierungen zu verhindern. Um die Lösung der Rutinoside zu gewährleisten, kön-
nen zusätzlich weitere Lösungsmittel zugegeben werden. Vorzugsweise wird das erfindungsgemäße Verfahren in Gegenwart eines Lösungsmittelgemisches aus Wasser und mindestens einem organischen Lösungsmittel durchgeführt.The rutinoside to be reacted is usually fed into the reactor, for example a column, such as a fixed bed column, in the form of a solution or suspension. If the reactor is a fixed bed reactor, the rutinoside solution should be completely free of solids. It is advantageous to pre-dissolve the rutinoside with the solvent in a tank, preferably with stirring and / or heating, in order to achieve optimal solubility. If necessary, pre-filtration of the solution can also be performed to remove any solids. The solvent is preferably an aqueous system to ensure enzyme activity and to prevent possible denaturation. To ensure the solution of the rutinosides, NEN additional solvents are added. The process according to the invention is preferably carried out in the presence of a solvent mixture of water and at least one organic solvent.
Das oder die zusätzlich vorhandenen organischen Lösungsmittel umfassen sowohl organische Lösungsmittel, die mit Wasser mischbar sind, als auch organische Lösungsmittel, die nicht mit Wasser mischbar sind.The organic solvent or solvents additionally present include both organic solvents which are miscible with water and organic solvents which are not miscible with water.
Geeignete Lösungsmittel für das erfmdungsgemäße Verfahren sind Nitrile, wie Aceto- nitril, Amide, wie Dimethylformamid, Ester, wie Essigsäureester, insbesondere Essigsäuremethylester oder Essigsäureethylester, Alkohole, wie Methanol oder Ethanol, Ether, wie Tetrahydrofuran oder Methyl-tert-butylether und Kohlenwasserstoffe, wie Toluol. Bevorzugt wird das erfindungsgemäße Verfahren in Gegenwart eines oder mehrerer der organischen Lösungsmittel Essigsäureester, Methanol, Ethanol, Methyl-tert-butylether oder Toluol durchgeführt. Besonders bevorzugt wird das erfindungsgemäße Verfahren in Gegenwart eines oder mehrerer Essigsäureester, insbesondere in Gegenwart von Essigsäuremethylester, zusätzlich zu Wasser durchgeführt.Suitable solvents for the process according to the invention are nitriles, such as acetonitrile, amides, such as dimethylformamide, esters, such as acetic acid esters, in particular methyl acetate or ethyl acetate, alcohols, such as methanol or ethanol, ethers, such as tetrahydrofuran or methyl tert-butyl ether and hydrocarbons, such as Toluene. The process according to the invention is preferably carried out in the presence of one or more of the organic solvents acetic acid ester, methanol, ethanol, methyl tert-butyl ether or toluene. The process according to the invention is particularly preferably carried out in the presence of one or more acetic acid esters, in particular in the presence of methyl acetate, in addition to water.
Geeignete Volumenverhältnisse von Wasser zu organischem Lösungsmittel für das erfindungsgemäße Verfahren sind Verhältnisse von 1 :99 bis 99:1. Vorzugsweise wird das erfindungsgemäße Verfahren mit Volumenverhältnissen von Wasser zu organischem Lösungsmittel von 20:80 bis 80:20, insbesondere mit Volumenverhältnissen von 50:50 bis 70:30 durchgeführt.Suitable volume ratios of water to organic solvent for the process according to the invention are ratios from 1:99 to 99: 1. The process according to the invention is preferably carried out with volume ratios of water to organic solvent from 20:80 to 80:20, in particular with volume ratios from 50:50 to 70:30.
Die Menge an Rutinosid in dem Lösungsmittel bzw. Lösungsmittelgemisch für das erfindungsgemäße Verfahren ist abhängig von der Löslichkeit des Rutinosids in dem Lösungsmittel bzw. Lösungsmittelgemisch. Zur optimalen Durchführung des erfindungsgemäßen Verfahrens sollte das Rutinosid gut löslich sein. Deshalb wird vorzugsweise mit einer untersättigten Lösung gearbeitet. Üblicherweise beträgt die Menge an Rutinosid im Lösungsmittel bzw. Lösungsmittelgemisch 0,001 bis 5 g/l, vorzugsweise 0,05 bis2 g/l, noch bevorzugter 0,1 bis 1 ,5 g/l.
Das Verhältnis von Rutinosid zu Immobilisat bzw. Enzym hängt von der Lebensdauer des Enzyms in der Säule und seiner Aktivität in immobilisierter Form ab.The amount of rutinoside in the solvent or solvent mixture for the process according to the invention depends on the solubility of the rutinoside in the solvent or solvent mixture. In order to carry out the process according to the invention optimally, the rutinoside should be readily soluble. It is therefore preferable to work with an undersaturated solution. The amount of rutinoside in the solvent or solvent mixture is usually 0.001 to 5 g / l, preferably 0.05 to 2 g / l, more preferably 0.1 to 1.5 g / l. The ratio of rutinoside to immobilizate or enzyme depends on the life of the enzyme in the column and its activity in immobilized form.
Die Reaktion wird üblicherweise bei einer Temperatur von 15 bis 80°C durchgeführt. Bevorzugt ist eine Temperatur von 30 bis 60°C, insbesondere ist eine Temperatur von 40 bis 50°C von Vorteil, um einer möglichen Zerstörung des Enzyms entgegenzuwirken und gleichzeitig eine hohe Löslichkeit des Rutinosids zu gewährleistenThe reaction is usually carried out at a temperature of 15 to 80 ° C. A temperature of 30 to 60 ° C. is preferred, in particular a temperature of 40 to 50 ° C. is advantageous in order to counteract possible destruction of the enzyme and at the same time to ensure a high solubility of the rutinoside
Wenn die Reaktionstemperatur zu niedrig ist, läuft die Reaktion durch absinkende Enzymaktivität mit einer unangemessenen langsamen Reaktionsgeschwindigkeit ab. Außerdem verringert sich die Löslichkeit des Rutinosids derart, dass unnötig hohe Lösungsmittelmengen notwendig werden. Wenn die Reaktionstemperatur dagegen zu hoch ist, wird das Enzym, welches ein Protein ist, denaturiert und somit deaktiviert.If the reaction temperature is too low, the reaction proceeds at an inappropriately slow reaction rate due to decreasing enzyme activity. In addition, the solubility of the rutinoside is reduced in such a way that unnecessarily high amounts of solvent are necessary. On the other hand, if the reaction temperature is too high, the enzyme, which is a protein, is denatured and thus deactivated.
Wenn das erfindungsgemäße Verfahren bei einer erhöhten Temperatur durchgeführt werden soll, kann der Reaktor mit einer Temperiereinrichtung versehen werden. Übliche Temperiereinrichtungen beinhalten ein Heizschlangensystem oder Doppelmantel. Es ist weiterhin von Vorteil, wenn das umzusetzende Rutinosid und insbesondere die Rutino- sidlösung vor Eintritt in den Reaktor temperiert wird. Zu diesem Zweck ist es üblich, dass die Rutinosidlösung aus einem temperierten Tank, der auf die für die Reaktion gewünschte Temperatur eingestellt ist, gespeist wird. Alternativ kann die einzuspeisende Lösung durch einen beheizbaren Schlauch geleitet werden, um die gewünschte Temperatur vor Eintritt in den Reaktor einzustellen. Durch die Temperierung können ebenfalls mögliche Auskristallisierungen des Rutinosids vermieden werden.If the process according to the invention is to be carried out at an elevated temperature, the reactor can be provided with a temperature control device. Common temperature control devices include a heating coil system or double jacket. It is furthermore advantageous if the rutinoside to be reacted and in particular the rutinoside solution is tempered before entering the reactor. For this purpose, it is common for the rutinoside solution to be fed from a temperature-controlled tank which is set to the temperature desired for the reaction. Alternatively, the solution to be fed can be passed through a heatable hose in order to set the desired temperature before entering the reactor. Possible tempering out of the rutinoside can also be avoided by the temperature control.
Geeignete pH-Werte für das erfindungsgemäße Verfahren sind pH-Werte zwischen 3 und 8. Vorzugsweise wird das erfindungsgemäße Verfahren, bei pH-Werten von 3 bis 7 durchgeführt, insbesondere bei pH-Werten von 3 bis 6. Weiterhin bevorzugte pH-Werte können jedoch je nach verwendetem Enzym innerhalb der gegebenen Grenzen variieren. Beispielsweise sind pH-Werte von 3,8 bis 4,3 bei Verwendung des Enzyms Hesperidinase ganz außerordentlich bevorzugt.
Vorzugsweise wird das Verfahren derart ausgeführt, dass der pH-Wert mit Hilfe eines Puffersystems eingestellt wird. Prinzipiell können alle gängigen Puffersysteme, die zur Einstellung der obengenannten pH-Werte geeignet sind, verwendet werden. Bevorzugt wird jedoch wässriger Citratpuffer verwendet.Suitable pH values for the method according to the invention are pH values between 3 and 8. The method according to the invention is preferably carried out at pH values from 3 to 7, in particular at pH values from 3 to 6. However, further preferred pH values can be used vary within the given limits depending on the enzyme used. For example, pH values from 3.8 to 4.3 are extremely preferred when using the enzyme hesperidinase. The method is preferably carried out in such a way that the pH is adjusted with the aid of a buffer system. In principle, all common buffer systems that are suitable for setting the above-mentioned pH values can be used. However, aqueous citrate buffer is preferably used.
Das Rutinosidgemisch, das in Form einer Lösung oder einer Suspension vorliegen kann, wird in den Reaktor, der das Immobilisat enthält, gegeben, um die enzymatische Hydrolyse durchzuführen. Diese Reaktion kann batchweise, d.h. diskontinuierlich, oder kontinuierlich durchgeführt werden.The rutinoside mixture, which may be in the form of a solution or a suspension, is added to the reactor containing the immobilizate to carry out the enzymatic hydrolysis. This reaction can be batch, i.e. be carried out batchwise or continuously.
Wird die Reaktion diskontinuierlich durchgeführt, dann wird üblicherweie eine Rutinosidsuspension in den Reaktor gegeben. Der Umsatzgrad wird durch die Menge an Rutinosid und Immobilisat bestimmt. Üblicherweise liegt das Verhältnis von Rutinosid zu Immobilisat bei 100:1 bis 1:1000, vorzugsweise 10:1 bis 1:100, noch bevorzugter 1:1 bis 1 :20. Das Verhältnis des Immobilisats zum Gesamtvolumen der Suspension beträgt üblicherweise 1:1000 bis 1:1 , vorzugsweise 1:100 bis 1:2, noch bevorzugter 1 :50 bis 1:5. Die Verweilzeit im Reaktor beträgt normalerweise 1h bis 10 Tage, vorzugsweise 8 h bis 4 Tage, noch bevorzugter 1 bis 2 Tage.If the reaction is carried out batchwise, a rutinoside suspension is usually added to the reactor. The degree of conversion is determined by the amount of rutinoside and immobilizate. The ratio of rutinoside to immobilizate is usually 100: 1 to 1: 1000, preferably 10: 1 to 1: 100, more preferably 1: 1 to 1:20. The ratio of the immobilizate to the total volume of the suspension is usually 1: 1000 to 1: 1, preferably 1: 100 to 1: 2, more preferably 1:50 to 1: 5. The residence time in the reactor is normally 1 hour to 10 days, preferably 8 hours to 4 days, more preferably 1 to 2 days.
Wird die Reaktion kontinuierlich durchgeführt, dann wird eine Rutinosidlösung üblicherweise permanent und kontinuierlich mit einer geeigneten Pumpe durch den Reaktor, vorzugsweise eine Säule oder MSFB-Reaktor, gefördert. Durch die entsprechende Einstellung der Fließgeschwindigkeit kann ein beliebig hoher Umsatzgrad erreicht werden. Normalerweise wird mit einer Fließgeschwindigkeit von 0,001 bis 1 mm/s, bezogen auf den leeren Rohrquerschnitt der Säule, gearbeitet.If the reaction is carried out continuously, a rutinoside solution is usually continuously and continuously conveyed through the reactor, preferably a column or MSFB reactor, using a suitable pump. By setting the flow rate accordingly, any desired degree of conversion can be achieved. Normally, a flow rate of 0.001 to 1 mm / s, based on the empty pipe cross-section of the column, is used.
Die Aktivität des Enzyms in dem System nimmt erfahrungsgemäß mit der Zeit ab. Es ist deshalb notwendig, dass das Immobilisat in regelmäßigen Abständen ganz oder teilweise erneuert wird. Um einen Aktivitätsverlust der Enzyme auszugleichen, ist es vorteilhaft, den Umsatzgrad durch UV- oder Rl-Detektion auszuwerten, so dass bei einer Änderung der Zusammensetzung eine Steuerung über die Pumpenleistung der Änderung entgegensteuert.
Nach Austritt der Reaktionslösung kann das erhaltene Produkt getrennt werden. Nach beendeter Reaktion besteht das Reaktionsgemisch hauptsächlich aus Lösungsmittel, nichtumgesetztem Rutinosid, Rhamnose, dem gewünschten monoglycosidierten Flavo- noid und möglicherweise weiteren Zusätzen, wie Puffer. Üblicherweise fällt das mo- noglycosidierte Flavonoid aus, sobald die Löslichkeitsgrenze erreicht ist und reichert sich nach und nach als Feststoff an.Experience has shown that the activity of the enzyme in the system decreases over time. It is therefore necessary for the immobilisate to be replaced in whole or in part at regular intervals. In order to compensate for a loss of activity of the enzymes, it is advantageous to evaluate the degree of conversion by UV or RI detection, so that when the composition is changed, control via the pump output counteracts the change. After the reaction solution has emerged, the product obtained can be separated. After the reaction has ended, the reaction mixture consists mainly of solvent, unreacted rutinoside, rhamnose, the desired monoglycosidated flavonoid and possibly other additives, such as buffers. The monoglycosidated flavonoid usually precipitates as soon as the solubility limit is reached and gradually accumulates as a solid.
Bei einer diskontinuierlichen Fahrweise mit magnetischen Trägermaterialien kann das immobilisierte Enzym nach beendeter Reaktion auf einfache Weise mit Hilfe einer magnetischen Abscheidevorrichtung von der Produktsuspension abgetrennt werden. Im Labormaßstab kann dazu ein starker Permanentmagnet in Plattenform verwendet werden. Es gibt aber auch größere Separatoren, die für verschiedenste industrielle Anwendungen entwickelt wurden und meist nach dem HGMS-Prinzip arbeiten (High gradient magnetic Separation). Eine solche Anlage kann z.B. aus einem senkrecht stehenden Strömungsrohr bestehen, das eine Packung aus feinen Edelstahldrähten enthält. Mittels geeignet angeordneter elektromagnetischer Spulen werden an den Drähten hohe Magnetfeldgradienten erzeugt und dadurch eine sehr effektive Abtrennung selbst kleinster Partikel im Größenbereich von Nanometem erzielt. Sofern die Magnetpartikel superpa- ramagnetisch sind, d.h. in Abwesenheit eines äußeren Magnetfeldes keine Remanenzmagnetisierung aufweisen, können sie nach Abschalten des Magnetfeldes durch wiederholtes Spülen mit Wasser problemlos wieder vollständig aus dem Separator entfernt werden.In the case of a discontinuous procedure with magnetic carrier materials, the immobilized enzyme can be separated from the product suspension in a simple manner with the aid of a magnetic separation device after the reaction has ended. A strong permanent magnet in plate form can be used on a laboratory scale. However, there are also larger separators that were developed for a wide variety of industrial applications and mostly work according to the HGMS principle (high gradient magnetic separation). Such a system can e.g. consist of a vertical flow tube, which contains a package of fine stainless steel wires. By means of suitably arranged electromagnetic coils, high magnetic field gradients are generated on the wires and a very effective separation of even the smallest particles in the nanometer range is achieved. If the magnetic particles are superparamagnetic, i.e. In the absence of an external magnetic field, there is no remanence magnetization, after switching off the magnetic field, it can be completely removed again from the separator by repeated rinsing with water.
Die Isolierung des gewünschten Reaktionsprodukts erfolgt nach gängigen Methoden bei üblicher Aufarbeitung.The desired reaction product is isolated by customary methods with customary workup.
Vorzugsweise wird das Produkt bei Aufkonzentrierung ausgefällt. Wenn das Lösungsmittel ein Lösungsmittelgemisch, enthaltend mindestens ein organisches Lösungsmittel, enthält, ist es bevorzugt, dass das organische Lösungsmittel unter vermindertem Druck abdestilliert wird. Das auskristallisierende monoglycosidierte Flavonoid wird üblicherweise vom restlichen Reaktionsgemisch durch eine Fest/Flüssigtrennung, wie Absaugen bzw. Filtration unter vermindertem Druck oder durch Abschleudern der ausgefallenen
Kristalle, getrennt. Anschließend wird der Feststoff gewaschen, vorzugsweise mit Wasser und danach getrocknet.The product is preferably precipitated when concentrated. If the solvent contains a mixed solvent containing at least one organic solvent, it is preferred that the organic solvent be distilled off under reduced pressure. The crystallizing monoglycosidated flavonoid is usually separated from the remaining reaction mixture by solid / liquid separation, such as suction or filtration under reduced pressure or by centrifuging off the precipitated Crystals, separated. The solid is then washed, preferably with water and then dried.
Alternativ kann zunächst der gesamte Reaktorinhalt abfiltriert werden. Der Filterkuchen, der das Produkt enthält, wird anschließend mit einem Lösungsmittel oder einem Puffer- Lösungsmittelgemisch behandelt, in dem das Produkt löslich ist. Dabei wird das Reaktionsprodukt aus dem Filterkuchen extrahiert.Alternatively, the entire reactor contents can first be filtered off. The filter cake containing the product is then treated with a solvent or a buffer-solvent mixture in which the product is soluble. The reaction product is extracted from the filter cake.
Bei einer diskontinuierlichen Fahrweise bleibt der in diesem Gemisch der unlösliche Katalysator, das Immobilisat, zurück. Dafür ist es notwendig, dass sich das Lösungsmittel oder das Puffer-Lösungsmittelgemisch nicht nachteilig auf das Enzym auswirken. Es hat sich gezeigt, dass das immobilisierte Enzym, z.B. Naringinase oder Hesperidina- se, in bestimmten Lösungsmittel-Puffergemischen oder unter mäßig alkalischen Bedingungen zwar keinerlei oder nur einen Bruchteil der ursprünglichen Aktivität besitzt, dass aber die Aktivität praktisch vollständig wiederhergestellt werden kann, wenn das Enzym anschließend sorgfältig mit einem Puffer im pH-Bereich 4-6 gespült wird; es handelt sich also um einen vorübergehenden Aktivitätsverlust, nicht um eine irreversible Denaturierung des Enzyms.In the case of a batchwise procedure, the catalyst, the immobilisate, which is insoluble in this mixture, remains. For this it is necessary that the solvent or the buffer-solvent mixture do not have an adverse effect on the enzyme. It has been shown that the immobilized enzyme, e.g. Naringinase or hesperidinase, in certain solvent-buffer mixtures or under moderately alkaline conditions, has no or only a fraction of the original activity, but the activity can be practically completely restored if the enzyme is subsequently carefully mixed with a buffer in the pH range 4 -6 is rinsed; it is therefore a temporary loss of activity, not an irreversible denaturation of the enzyme.
Für diese Prozedur sehr gut geeignete Extraktionsmittel sind Tetrahydrofuran-Puffer- Gemische - vorzugsweise mit einem Tetrahydrofuran-Anteil von 10-25%, insbesondere auch bei leicht erhöhter Temperatur. Andere geeignete Extraktionsmittelkomponenten sind z.B. 1-Propanol, 2-Propanol, 1 ,4-Dioxan und Methylacetat. Das Produkt lässt sich aus dem Extrakt sehr leicht zurückgewinnen, indem man das Lösungsmittel unter reduziertem Druck abdestilliert und die wässrige Produktlösung anschließend auf 0 bis 10°C abkühlt. Das Reaktionsprodukt kristallisiert aus der Mutterlauge in sehr hoher Reinheit aus.Extraction agents which are very suitable for this procedure are tetrahydrofuran buffer mixtures - preferably with a tetrahydrofuran content of 10-25%, in particular also at a slightly elevated temperature. Other suitable extractant components are e.g. 1-propanol, 2-propanol, 1, 4-dioxane and methyl acetate. The product can be very easily recovered from the extract by distilling off the solvent under reduced pressure and then cooling the aqueous product solution to 0 to 10 ° C. The reaction product crystallizes out of the mother liquor in very high purity.
Alternativ zu Lösungsmittel/Puffer-Gemischen kann auch eine verdünnte Ammoniakoder Sodalösung als Extraktionsmittel eingesetzt werden, da das Reaktionsprodukt phenolische OH-Gruppen besitzt, welche bereits in schwach basischem Medium deprotoniert werden; das Anion des Reaktionsproduktes ist vergleichsweise gut löslich, allerdings auch sehr oxidationsempfindlich, was sich durch eine allmähliche Verfärbung
des Extraktes von gelb nach braun bemerkbar macht. Daher sollte bei dieser Variante zügig gearbeitet werden, d.h. die Extraktion sollte innerhalb von 10 min bis 6 h, vorzugsweise 20 min bis 2 h abgeschlossen sein. Vorzugsweise ist unter einer Schutzgasatmosphäre zu arbeiten.As an alternative to solvent / buffer mixtures, a dilute ammonia or soda solution can also be used as the extraction agent, since the reaction product has phenolic OH groups which are already deprotonated in a weakly basic medium; the anion of the reaction product is comparatively readily soluble, but is also very sensitive to oxidation, which is the result of a gradual discoloration of the extract from yellow to brown. Therefore, this variant should be operated quickly, ie the extraction should be completed within 10 minutes to 6 hours, preferably 20 minutes to 2 hours. It is preferable to work in a protective gas atmosphere.
Auch durch die Behandlung mit schwach basischen Extraktionsmitteln, wie wässrigen Lösungen von Alkali- oder Ammoniumsalzen der Essigsäure, Oxalsäure, Citronensäure, Phosphorsäure, Borsäure oder Kohlensäure, oder wässrigen Lösungen von Alkylami- nen, Piperidin oder Pyridin, geht die Enzymaktivität nicht verloren. Durch Ansäuern des Extrakts und Abkühlung auf 0-10°C kann das Reaktionsprodukt wieder ausgefällt werden.The enzyme activity is not lost even by treatment with weakly basic extraction agents, such as aqueous solutions of alkali or ammonium salts of acetic acid, oxalic acid, citric acid, phosphoric acid, boric acid or carbonic acid, or aqueous solutions of alkylamines, piperidine or pyridine. The reaction product can be precipitated again by acidifying the extract and cooling to 0-10 ° C.
Die Reinheit des erhaltenen monoglycosidierten Flavonoids bei Einsatz von reinem Rutinosid ist normalerweise größer als 94%. Zur weiteren Reinigung kann das Endprodukt beispielsweise aus geeigneten Lösungsmitteln umkristallisiert werden, z.B. aus Wasser oder aus Lösungsmittelgemischen, bestehend aus Toluol und Methanol oder Wasser und Essigsäuremethylester.The purity of the monoglycosidated flavonoid obtained using pure rutinoside is normally greater than 94%. For further purification, the end product can, for example, be recrystallized from suitable solvents, e.g. from water or from solvent mixtures consisting of toluene and methanol or water and methyl acetate.
Die anfallende Menge an Lösungsmittel nach der Reaktion wird vorzugsweise zurückgewonnen, um die Wirtschaftlichkeit des erfindungsgemäßen Verfahrens zu gewährleisten. Diese Rezirkulation erfolgt üblicherweise kontinuierlich und automatisiert. Hierfür bieten sich handelsübliche Verdampferanlagen mit entsprechender Steuerung an. Handelt es sich bei dem einzusetzenden Lösungsmittel um ein Lösungsmittelgemisch aus Wasser und mindestens einem organischen Lösungsmittel, ist es in der Regel nicht möglich, das Destillat sofort wieder für das Verfahren einzusetzen, da sich die Lösungsmittelverhältnisse durch das Abdestillieren des organischen Lösungsmittels verändert haben. Durch automatische Qualitätsmessung und Korrektur kann, das gewünschte Lösungsmittelverhältnis dabei wiederhergestellt werden und das Lösungsmittel somit rückgeführt werden.The amount of solvent obtained after the reaction is preferably recovered in order to ensure the economy of the process according to the invention. This recirculation is usually continuous and automated. Commercial evaporator systems with appropriate controls are available for this. If the solvent to be used is a solvent mixture of water and at least one organic solvent, it is generally not possible to immediately use the distillate again for the process, since the solvent ratios have changed as a result of the organic solvent being distilled off. The desired solvent ratio can be restored by automatic quality measurement and correction and the solvent can thus be returned.
Weiterhin können bei der Aufkonzentrierung Membranprozesse oder Nanofiltration durchgeführt werden. Bei diesen Verfahren wird das Lösungsmittelgemisch ohne Änderung der Zusammensetzung abgetrennt.
Die folgenden Beispiele sollen die vorliegende Erfindung verdeutlichen. Sie sind jedoch keinesfalls als beschränkend zu betrachten.Furthermore, membrane processes or nanofiltration can be carried out during the concentration. In this process, the solvent mixture is separated without changing the composition. The following examples are intended to illustrate the present invention. However, they are in no way to be regarded as restrictive.
Beispiel 1example 1
Immobilisierung des Enzyms Hesperidinase auf einem KieselgelträgerImmobilization of the enzyme hesperidinase on a silica gel support
1) Trägeroberflächenkonditionierung zur Immobilisierung1) Carrier surface conditioning for immobilization
1.1) Eigenschaften des Träqermaterials Kieselgel LiChrospher Durchmesser = 15 - 40 μm Porendurchmesser = 300 A Partikeloberfläche = 80 m2/g Porenvolumen = 0,73 ml/g Dichte = 2 g/ml1.1) Properties of the carrier material silica gel LiChrospher diameter = 15 - 40 μm pore diameter = 300 A particle surface = 80 m 2 / g pore volume = 0.73 ml / g density = 2 g / ml
1.2) Kieselqel-Aktivierung1.2) Silica activation
250 g Kieselgel werden mit genügend HCI (7%) in einer 1 L-Flasche vermengt und über Nacht stehen gelassen, um das Kieselgel zu befeuchten.250 g of silica gel are mixed with sufficient HCI (7%) in a 1 L bottle and left overnight to moisten the silica gel.
Anschließend wird die Kieselgelsuspension mit VE-Wasser chloridfrei gewaschen. Dazu muss man nach jeder Waschung den Überstand mit Salpetersäure und Silbernitrat testen. Die Waschung wird wegen der Eigenschaften der Kieselgelpartikel auf einem ca. 24 cm-Durchmesser-Keramiktrichter durchgeführt.The silica gel suspension is then washed chloride-free with demineralized water. To do this, you have to test the supernatant with nitric acid and silver nitrate after each wash. Because of the properties of the silica gel particles, the washing is carried out on an approximately 24 cm diameter ceramic funnel.
1.3) Oberflächenmodifiaktion mit Aminoqruppen1.3) Surface modification with amino groups
Das säurebehandelte Kieselgel wird in einem 2 L-Dreihalskolben, der mit einem Rückflußkühler sowie einem Tropftrichter ausgestattet ist, zusammen mit genügend Wasser gemischt, so dass es rührfähig wird. Bei Raumtemperatur und guter Durchmi-
schung tropft man 1 mmol/g Träger des 3-Aminopropyltrimethoxysilans (für 250 g Kieselgel braucht man 135 ml Lösung) mit einer Geschwindigkeit von ca. 5 Tropfen/s zu der Kieselgelsuspension zu. Anschließend wird die Suspension 2 Stunden bei 90°C gerührt. Danach wird die Suspension mit Eis gekühlt.The acid-treated silica gel is mixed with enough water in a 2 L three-necked flask equipped with a reflux condenser and a dropping funnel, so that it becomes stirrable. At room temperature and good mixing 1 mmol / g carrier of the 3-aminopropyltrimethoxysilane (for 250 g of silica gel, 135 ml of solution is required) is added dropwise to the silica gel suspension at a rate of about 5 drops / s. The suspension is then stirred at 90 ° C. for 2 hours. The suspension is then cooled with ice.
Der Überstand muss mittels pH-Wert Messung auf mögliche Reste von 3-Amino- propyltrimethoxysilan überprüft werden. Die Beadsuspension wird mit VE-Wasser so lange gewaschen, bis der pH-Wert konstant bleibt.The supernatant must be checked for possible residues of 3-aminopropyltrimethoxysilane by means of pH measurement. The bead suspension is washed with demineralized water until the pH remains constant.
14) Glutardialdehvdbelequng14) Glutardialdehyde coverage
Zu der erhaltenen Kieselgelsuspension wird Glutardialdehyd (GDA) in einer Konzentration von 1 mmol/g Träger (für 250 g Träger braucht man 13 ml 50%-ige GDA-Lösung) zugegeben. Die Suspension (plus etwas Wasser) wird 2 Stunden bei Raumtemperatur in einer 1 L-Flasche gerollt. Am Anfang ist die Suspension gelb gefärbt, und am Ende wird sie dunkelrot.Glutardialdehyde (GDA) is added to the silica gel suspension obtained in a concentration of 1 mmol / g of carrier (13 ml of 50% GDA solution are required for 250 g of carrier). The suspension (plus some water) is rolled in a 1 L bottle at room temperature for 2 hours. At the beginning the suspension is colored yellow and at the end it turns dark red.
Anschließend wird der Überstand von jedem Waschen durch eine Fällungsreaktion mit Dinitrophenylhydrazin auf Glutardialdehydreste überprüft. Die Suspension wird sorgfältig gewaschen, bis der Test negativ ist.The supernatant from each wash is then checked for residues of glutardialdehyde by a precipitation reaction with dinitrophenylhydrazine. The suspension is washed carefully until the test is negative.
2) Immobilisierung2) Immobilization
2.1) Hesperidinase2.1) hesperidinase
Erste ProteinzugabeFirst addition of protein
3,8 g Hesperidinase werden in 500 ml Citrat/Phosphatpuffer (pH = 6,0) gerührt. Zum besseren Lösen werden 300 μl Tenside (Tween 20) zugegeben. Anschließend wird die Enzymlösung filtriert.
In einer 1 L-Flasche werden ca. 230 g der unter 1.4) erhaltenen Kieselgelsuspension mit der Enzymlösung vermengt. Anschließend wird die Enzym-Trägersuspension ca. 40 Stunden bei Raumtemperatur gerollt.3.8 g of hesperidinase are stirred in 500 ml of citrate / phosphate buffer (pH = 6.0). 300 μl of surfactants (Tween 20) are added for better dissolution. The enzyme solution is then filtered. In a 1 L bottle, approx. 230 g of the silica gel suspension obtained under 1.4) are mixed with the enzyme solution. The enzyme carrier suspension is then rolled at room temperature for about 40 hours.
Zweite ProteinzugabeSecond protein addition
ca. 0,76 g Hesperidinase (Amano) werden in 120 ml Citrat/Phosphatpuffer (pH = 6,0) mit 60 μl Tensiden gerührt und später filtriert.Approx. 0.76 g hesperidinase (Amano) are stirred in 120 ml citrate / phosphate buffer (pH = 6.0) with 60 μl surfactants and later filtered.
Die Enzymlösung wird in die vorstehend genannte 1 L-Flasche gegossen, und die Enzymlösung wird bei Raumtemperatur gerollt.The enzyme solution is poured into the 1 L bottle mentioned above, and the enzyme solution is rolled at room temperature.
2.2) BSA (für den Trennunqstest)2.2) BSA (for the separation test)
0,3 Biomex BSA (Rinderserumalbumin-Pulver) werden in 100 ml Citrat/Phosphatpuffer (pH = 6,0) gerührt. In einer 0,5 L-Flasche werden ca. 20 g der unter 1.4) erhaltenen Kieselgelsuspension mit der Proteinlösung vermengt, und 60 μl ProClin300 werden zugegeben.0.3 Biomex BSA (bovine serum albumin powder) is stirred in 100 ml citrate / phosphate buffer (pH = 6.0). Approx. 20 g of the silica gel suspension obtained under 1.4) are mixed with the protein solution in a 0.5 L bottle, and 60 μl ProClin300 are added.
3) Proteinmengen- und Aktivitätsbestimmung3) Protein quantity and activity determination
3.1) Proteinmenge (mg Protein/ml)3.1) Amount of protein (mg protein / ml)
Der Proteingehalt einer Lösung wird mittels Bradford-Test bestimmt. Es wird das Stan- dard-Assay durchgeführt. Dabei werden 20 μl Probe in 1 ml Bradford-Farbstoff-Reagenz (1 :5 verdünnt) gemischt und nach 15 min im Photometer bei 595 nm gemessen.The protein content of a solution is determined using the Bradford test. The standard assay is performed. 20 μl sample are mixed in 1 ml Bradford dye reagent (diluted 1: 5) and measured after 15 min in a photometer at 595 nm.
Bei sehr geringen Proteinkonzentrationen muss das Microassay durchgeführt werden. Dabei werden 0,8 ml Probe in 0,2 ml Bradford-Farbstoff-Reagenz (konzentriert) gemischt und nach 15 min im Photometer bei 595 nm gemessen.
3.2) AktivitätThe microassay must be carried out at very low protein concentrations. 0.8 ml of sample are mixed in 0.2 ml of Bradford dye reagent (concentrated) and measured after 15 min in a photometer at 595 nm. 3.2) Activity
Die Aktivität einer Lösung wird durch die Reaktion mit einem Ersatzsubstrat gemessen. Für jede Probe nimmt man:The activity of a solution is measured by the reaction with a replacement substrate. For each sample one takes:
88 μl Citrat/Phosphatpuffer (pH = 4,0) 100 μl Probe 20 μl Probe88 ul citrate / phosphate buffer (pH = 4.0) 100 ul sample 20 ul sample
20 μl Ersatzsubstrat: p-Nitrophenyl-α-L-Rhamnosid (Rhamnosidase-Aktivität) p-Nitrophenyl- -L-Glucosid (Glucosidase-Akivität)20 μl replacement substrate: p-nitrophenyl-α-L-rhamnoside (rhamnosidase activity) p-nitrophenyl- -L-glucoside (glucosidase activity)
Diese 1 ml Lösung wird in einem Eppendorfreaktionsgefäß vermengt. Nach einer Inkubationszeit von je 2 und 5 min bei 40°C im Schüttler, werden je 100 μl des Reaktionsgemisches mit 1 ml 1 M-Sodalösung vermengt. Dann wird die Konzentration von p-Nitrophenol im Photometer bei 400 nm gemessen. Die Aktivität errechnet sich durch Konzentrationsänderung von p-Nitrophenol pro Zeit.This 1 ml solution is mixed in an Eppendorf reaction vessel. After an incubation time of 2 and 5 min at 40 ° C in a shaker, 100 μl of the reaction mixture are mixed with 1 ml of 1 M soda solution. Then the concentration of p-nitrophenol is measured in the photometer at 400 nm. The activity is calculated by changing the concentration of p-nitrophenol per time.
Die Aktivität eines Enzyms wird in Units (U) (= μmol umgesetztes Substrat je Minute) angegeben.
The activity of an enzyme is given in units (U) (= μmol converted substrate per minute).
4) Ergebnisse4) Results
PROTEINGEHALT und AKTIVITÄTSWERTE (FREIE HESPERIDINASE)
PROTEIN CONTENT and ACTIVITY VALUES (FREE HESPERIDINASE)
1 HespO: erste Proteinzugabe Hespl : zweite Proteinzugabe Proben 1-6: Überstand 1 HespO: first protein addition Hespl: second protein addition Samples 1-6: supernatant
PROTEINGEHALT und AKTIVITÄTSWERTE (IMMOBILISIERTE HESPERIDINASE)PROTEIN CONTENT and ACTIVITY VALUES (IMMOBILIZED HESPERIDINASE)
Proteingehalt des Trägers ≤ 1 ,045 g gebundenes ProteinProtein content of the carrier ≤ 1, 045 g bound protein
Auf den Träger sind 4,7 mg Protein/g TrägerThere are 4.7 mg protein / g carrier on the carrier
2% des zugegebenen Proteins wurden nicht gebunden
Beispiel 22% of the added protein was not bound Example 2
Herstellung von Isoquercetin aus Rutin durch enzymatische Hydrolyse unter Verwendung eines Immobilisats.Preparation of isoquercetin from rutin by enzymatic hydrolysis using an immobilizate.
In einem heizbaren 4,5 m3-Rührkessel (1) werden 3200 I VE-Wasser und 800 I 1-Propanol vorgelegt. Über die Dampfzufuhr (2) wird das Gemisch auf ca. 50-60°C erwärmt. Unter starkem Rühren wird der Lösung 8000 g Rutin, DAB zugegeben. Die Mischung wird solange gerührt, bis sich das Rutin vollständig gelöst hat. Über eine Kreislaufpumpe und ein in die Leitung montiertes pH-Meter (3a) wird anschließend der pH- Wert kontrolliert und ggf. auf pH 4,0 - 4,5 eingestellt (mit H3P04 und NaOH). Über das manuelle Ventil (4) kann auch zur Kontrolle und Konzentrationsbestimmung eine Probe entnommen werden.3200 l of demineralized water and 800 l of 1-propanol are placed in a heatable 4.5 m 3 stirred tank (1). The mixture is heated to approx. 50-60 ° C via the steam supply (2). 8000 g of rutin, DAB are added to the solution with vigorous stirring. The mixture is stirred until the rutin has completely dissolved. The pH value is then checked using a circulating pump and a pH meter (3a) installed in the line and adjusted to pH 4.0 - 4.5 if necessary (with H 3 P0 4 and NaOH). A sample can also be taken via the manual valve (4) for checking and determining the concentration.
Zum Start der Reaktion wird die Lösung über einen Beutelfilter (5) und einen Kerzenfilter (6) der Kolbendosierpumpe (7) zugeführt. Der Beutelfilter hat dabei die Aufgabe, die größere Menge an ungelösten Bestandteilen aufzuhalten, während der Kerzenfilter die Lösung mit einer Feinheit von 0,2 μm reinigt.At the start of the reaction, the solution is fed to the piston metering pump (7) via a bag filter (5) and a candle filter (6). The bag filter has the task of stopping the larger amount of undissolved components, while the candle filter cleans the solution with a fineness of 0.2 μm.
Die Kolbendosierpumpe (7) fördert die Lösung durch einen heizbaren Schlauch, welcher die Lösung über ein Thermometer auf eine Temperatur von 40°C am Eingang der Säule einstellt, mit einem Fluss von 1 l/min in die Säule (9) (100x400 mmj. Die Säule enthält 1,5 kg Immobilisat. Da der elektrisch beheizte Schlauch die Lösung nicht kühlen kann, ist die Temperatur im Rührkessel (1) so zu wählen, dass die Abkühlung auf dem Weg bis zur Pumpe bei maximalem Pumpenfluss bis 40°C führt.The piston metering pump (7) conveys the solution through a heatable hose, which adjusts the solution to a temperature of 40 ° C at the entrance of the column using a thermometer, with a flow of 1 l / min into the column (9) (100x400 mmj. The column contains 1.5 kg of immobilized material, since the electrically heated hose cannot cool the solution, the temperature in the stirred tank (1) must be selected so that cooling on the way to the pump leads to a maximum pump flow of up to 40 ° C.
Über ein manuelles Ventil (10) kann der Lösung nach Durchlaufen der Säule eine Probe entnommen werden, womit nochmals die Temperatur und der Umsatzgrad der Reaktion off-iine gemessen werden können. Ist der gemessene Umsatzgrad geringer als gefordert, wird die Flussrate der Pumpe entsprechend verringert.A sample can be taken from the solution after passing through the column via a manual valve (10), with which the temperature and the degree of conversion of the reaction can be measured off-line again. If the measured degree of conversion is lower than required, the flow rate of the pump is reduced accordingly.
Nach Durchlaufen der Säule ist die Reaktion vollständig abgelaufen, so dass die Lösung in das Auffanggefäß (11) geführt werden kann. Dort wird der Lösung über einen Kon-
densator (12) ca. 10-20% des Volumens entzogen. Dadurch wird der Gehalt an Propa- nol deutlich gesenkt, wodurch die Löslichkeit des Isoquercetins stark sinkt. Durch nachträgliche Abkühlung sinkt die Löslichkeit weiter, so dass das Produkt ausfällt und im Beutefilter (13) abgetrennt werden kann. Von dort wird es zur weiteren Trocknung in einen Trockenschrank (14) überführt. Die Mutterlauge und das abdestillierte Kondensat werden gemeinsam zum erneuten Ansatz in den Rührkessel (1) zurückgeführt.After passing through the column, the reaction is complete, so that the solution can be fed into the collecting vessel (11). There the solution is about 10-20% of the volume is removed from the capacitor (12). This significantly reduces the propanol content, which greatly reduces the solubility of the isoquercetin. Subsequent cooling further reduces the solubility, so that the product precipitates and can be separated in the prey filter (13). From there it is transferred to a drying cabinet (14) for further drying. The mother liquor and the distilled-off condensate are returned together in the stirred tank (1) for fresh preparation.
Beispiel 3Example 3
1 Modifizierung von Kieselgelpartikeln mit Aldehvdqruppen und Immobilisieruno von Naringinase auf diesen Partikeln1 Modification of silica gel particles with aldehyde groups and immobilization of naringinase on these particles
250 g Kieselgel (z.B. LiChrospher Sl 300, Merck, Darmstadt) wurden in einem verschließbaren Gefäß mit 400 ml 10%iger HCI übergössen, 10 min lang mit Ultraschall entgast, und 24 h bei RT stehengelassen. Anschließend wurde das Kieselgel abfiltriert und mit mehreren Litern demin. Wasser gewaschen, bis der pH-Wert >4.5 betrug und im Filtrat keine Chloridionen mehr nachweisbar waren (Tüpfelreaktion mit essigsaurer AgN03 -Lösung).250 g of silica gel (eg LiChrospher Sl 300, Merck, Darmstadt) were poured with 400 ml of 10% HCl in a closable vessel, degassed with ultrasound for 10 minutes and left to stand at RT for 24 hours. The silica gel was then filtered off and demineralized with several liters. Washed water until the pH was> 4.5 and chloride ions were no longer detectable in the filtrate (spot reaction with acetic acid AgN0 3 solution).
Anschließend wurde das säurebehandelte feuchte Kieselgel in einen 4 1 Dreihalskolben - ausgestattet mit KPG-Rührer, Rückflußkühler und 100 ml Tropftrichter - überführt und mit 3 I VE-Wasser aufgeschlämmt. Unter Rühren wurden über den Tropftrichter innerhalb 15 min 100 ml Aminopropyltrimethoxysilan (ABCR, Karlsruhe) zugegeben. Anschließend wurde die Suspension erwärmt und 90 min lang bei 90°C gerührt. Die abgekühlte Suspension wurde filtriert und achtmal mit jeweils 1 I demin. Wasser gewaschen.The acid-treated moist silica gel was then transferred to a 4 1 three-necked flask equipped with KPG stirrer, reflux condenser and 100 ml dropping funnel and slurried with 3 l of deionized water. With stirring, 100 ml of aminopropyltrimethoxysilane (ABCR, Karlsruhe) were added via the dropping funnel over the course of 15 minutes. The suspension was then heated and stirred at 90 ° C. for 90 minutes. The cooled suspension was filtered and eight times with 1 l of demin. Washed water.
Das aminoaktivierte Kieselgel wurde in einem 4 I Dreihalskolben - ausgestattet mit KPG-Rührer und 100 ml Tropftrichter - in 3 I mittels Ultraschall entgastem Wasser suspendiert; der pH-Wert wurde mit einigen Tropfen 2 M Essigsäure auf 8.0 gesenkt. Anschließend wurden 100 ml 50%ige Glutardialdehydlösung (Merck, Darmstadt) innerhalb 1 h zugetropft und die Suspension weitere 2,5 h bei RT gerührt. Das aktivierte Kieselgel wird erneut filtriert und so lange mit eiskaltem demin. Wasser gewaschen, bis im
Waschwasser kein Glutaraldehyd mehr nachgewiesen werden konnte (Tüpfelreaktion mit schwefelsaurer 2,4-Dinitrophenylhydrazin-Lösung).The amino-activated silica gel was suspended in 3 I using ultrasonically degassed water in a 4 I three-necked flask equipped with KPG stirrer and 100 ml dropping funnel; the pH was lowered to 8.0 with a few drops of 2 M acetic acid. Then 100 ml of 50% glutardialdehyde solution (Merck, Darmstadt) were added dropwise within 1 h and the suspension was stirred for a further 2.5 h at RT. The activated silica gel is filtered again and so long with ice-cold demin. Washed water until Wash water no more glutaraldehyde could be detected (spot reaction with sulfuric acid 2,4-dinitrophenylhydrazine solution).
Das mit Aldedydgruppen modifizierte Kieselgel wurde in 500 ml demin. Wasser in einem 4 I Kolben durch Rühren mit einem KPG-Rührer suspendiert. 13 g Naringinase (Sigma, Deisenhofen) wurden in 2,5 1 0,25 M Phosphatpuffer, pH 8,0, gelöst. Die Enzymlösung wurde in die Kieselgelsuspension gegeben und 96 h lang bei RT gerührt. Anschließend wurde das Immobilisat abfiltriert und mehrfach zunächst mit zunächst mit 0,2 M Natriumchloridlösung, dann mit 50 mlll Citratpuffer, pH 4,0, gewaschen. Die Rhamnosidaseakti- vität des Immobilisats wurde mit p-Nitrophenyl-L-α-rhamnopyranosid (Sigma, Deisenhofen) als Substrat nach der Methode von Kurosawa bestimmt; sie betrug 120 U/g.The silica gel modified with aldehyde groups was deminized in 500 ml. Water is suspended in a 4 liter flask by stirring with a KPG stirrer. 13 g of naringinase (Sigma, Deisenhofen) were dissolved in 2.5 1 0.25 M phosphate buffer, pH 8.0. The enzyme solution was added to the silica gel suspension and stirred at RT for 96 h. The immobilizate was then filtered off and washed several times, first with 0.2 M sodium chloride solution and then with 50 ml of citrate buffer, pH 4.0. The rhamnosidase activity of the immobilizate was determined using p-nitrophenyl-L-α-rhamnopyranoside (Sigma, Deisenhofen) as substrate by the Kurosawa method; it was 120 U / g.
2. Immobilisierung von Hesperidinase auf Eupergit™ C2. Immobilization of hesperidinase on Eupergit ™ C
50 g Eupergit (Röhm, Weiterstadt) wurden in einer verschraubbaren 500 ml Glasflasche mit 300 ml 0,8 M Kaliumphosphatpuffer, pH 8,5, vermischt und 30 min lang stehen gelassen. Anschließend wurden 5,0 g Hesperidinase (Amano) zugegeben und der Ansatz 120 h lang bei RT auf einem Rollenmischer bewegt. Das Eupergit wurde mittels einer Glasfritte abfiltriert und mehrmals zunächst mit 0,2 M Natriumchloridlösung, anschließend zweimal mit je 1 I 0,1 M Citratpuffer, pH 4,0, gewaschen. Die Rhamnosidaseakti- vität des Immobilisats wurde mit p-Nitrophenyl-L- -rhamnopyranosid (Sigma, Deisenhofen) als Substrat nach der Methode von Kurosawa bestimmt; sie betrug 15 U/g, bezogen auf trockenes Immobilisat, bzw. 4,2 U/g, bezogen auf feuchtes Immobilisat.50 g of Eupergit (Röhm, Weiterstadt) were mixed in a screw-on 500 ml glass bottle with 300 ml of 0.8 M potassium phosphate buffer, pH 8.5, and left to stand for 30 minutes. Then 5.0 g of hesperidinase (Amano) were added and the mixture was stirred for 120 hours at RT on a roller mixer. The Eupergit was filtered off by means of a glass frit and washed several times first with 0.2 M sodium chloride solution, then twice with 1 I 0.1 M citrate buffer, pH 4.0. The rhamnosidase activity of the immobilizate was determined using p-nitrophenyl-L- rhamnopyranoside (Sigma, Deisenhofen) as substrate by the Kurosawa method; it was 15 U / g, based on dry immobilizate, or 4.2 U / g, based on moist immobilisate.
3. Umsetzung von Rutin zu lsoguercetin mit auf Eupergit immobilisierter Hesperidinase im Rührkesselreaktor und anschließende Extraktion des Produkts mit einer Tetra- hvdrofuran/Puffer-Mischung3. Conversion of rutin to isoguercetin with hesperidinase immobilized on Eupergit in a stirred tank reactor and subsequent extraction of the product with a tetrahydrofuran / buffer mixture
In einem 2000 ml Rundkolben wurden 1000 ml 50 mM Citratpuffer, pH 4,0, 100 g (Feuchtgewicht) auf Eupergit immobilisierte Naringinase mit einer Aktivität von 4,2 U/g, sowie 10 g Rutin (Merck, Darmstadt) bei 40°C mit einem KPG-Rührer gerührt; der Grad des Umsatzes wurde laufend durch HPLC-Analyse bestimmt. Nach insgesamt 96 h
wurde der Reaktorinhalt über einen Büchnertrichter abfiltriert. Der Filterkuchen wurde wieder in den Rundkolben zurück gegeben und 30 min lang in einer Mischung aus 400 ml 50 mM Citratpuffer, pH 4,0, und 100 ml Tetrahydrofuran bei 40°C gerührt, wobei sich der größte Teil des Isoquercetins löste. Das Gemisch wurde warm filtriert und der Filterkuchen erneut 30 min lang mit 500 ml Puffer-Tetrahydrofuran-Ge isch extrahiert. Nach der Filtration wurden die beiden Isoquercetin-Extrakte mit dem ersten Filtrat vereinigt und das Tetrahydrofuran mit Hilfe eines Rotationsverdampfers entfernt. Um das Produkt vollständig auszufällen wurde die wässrige Isoquercetinlösung auf 4°C abgekühlt,. Nach Filtration und Trocknung im Exsikkator wurde eine Ausbeute von 5,8 g Produkt erhalten, das sich aus 98% Isoquercetin und 2% Rutin zusammensetzte.1000 ml of 50 mM citrate buffer, pH 4.0, 100 g (wet weight) immobilized on Eupergit with an activity of 4.2 U / g, and 10 g of rutin (Merck, Darmstadt) at 40 ° C. were placed in a 2000 ml round-bottomed flask stirred with a KPG stirrer; the degree of turnover was continuously determined by HPLC analysis. After a total of 96 h the reactor contents were filtered off via a Büchner funnel. The filter cake was returned to the round bottom flask and stirred for 30 minutes in a mixture of 400 ml of 50 mM citrate buffer, pH 4.0 and 100 ml of tetrahydrofuran at 40 ° C., with most of the isoquercetin dissolving. The mixture was filtered warm and the filter cake extracted again with 500 ml of buffer tetrahydrofuran mixture for 30 minutes. After filtration, the two isoquercetin extracts were combined with the first filtrate and the tetrahydrofuran was removed using a rotary evaporator. In order to completely precipitate the product, the aqueous isoquercetin solution was cooled to 4 ° C. After filtration and drying in a desiccator, a yield of 5.8 g of product was obtained, which was composed of 98% isoquercetin and 2% rutin.
Das feuchte Eupergit wurde einmal mit kaltem Tetrahydrofuran-Puffergemisch gewaschen, danach wiederholt mit 50 mM Citratpuffer, pH 4,0, bis der Geruch von Tetrahydrofuran nur noch schwach wahrzunehmen war. Die Aktivität des Enzyms betrug noch 3,6 U/g, das entspricht einem Aktivitätsverlust von 14%.The moist eupergite was washed once with cold tetrahydrofuran buffer mixture, then repeatedly with 50 mM citrate buffer, pH 4.0, until the smell of tetrahydrofuran was barely perceptible. The activity of the enzyme was still 3.6 U / g, which corresponds to an activity loss of 14%.
4. Umsetzung von Rutin zu Isoguercetin mit auf Eupergit immobilisierter Hesperidinase im Rührkesselreaktor und anschließende Extraktion des Produkts mit einer alkalischen Pufferlösung4. Conversion of rutin to isoguercetin with hesperidinase immobilized on Eupergit in a stirred tank reactor and subsequent extraction of the product with an alkaline buffer solution
In einem 2000 ml Rundkolben wurden 1000 ml 50 mM Citratpuffer, pH 4,0, 100 g auf Eupergit immobilisierte Naringinase mit einer Aktivität von 4,2 U/g (Feuchtgewicht), sowie 10 g Rutin (Merck, Darmstadt) bei 40°C mit einem KPG-Rührer gerührt; der Grad des Umsatzes wurde laufend durch HPLC-Analyse bestimmt. Nach insgesamt 96 h wurde der Reaktorinhalt über einen Büchnertrichter abfiltriert. Der feuchte Filterkuchen wurde wieder in den Rundkolben zurück gegeben und bei RT 5 min lang in 300 ml 50 mM Natriumcarbonatpuffer, pH 10,0, gerührt, wobei sich ein Teil des Isoquercetins mit intensiv gelber Farbe löste. Die Suspension wurde filtriert und der Filterkuchen sofort erneut mit Carbonatpuffer extrahiert. Nach insgesamt 7 Extraktionszyklen war das Eupergit weitgehend farblos und das Isoquercetin praktisch vollständig gelöst. Die Extrakte wurden vereinigt, mit verdünnter Salzsäure vorsichtig angesäuert, bis der pH ungefähr bei 3 lag und anschließend auf 4°C gekühlt. Nach Filtration und Trocknung im Exsikka-
tor wurde eine Ausbeute von 4,9 g Produkt erhalten, das sich aus 98% Isoquercetin und 2% Rutin zusammensetzte.1000 ml of 50 mM citrate buffer, pH 4.0, 100 g of naringinase immobilized on Eupergit with an activity of 4.2 U / g (wet weight) and 10 g of rutin (Merck, Darmstadt) at 40 ° C. were placed in a 2000 ml round-bottomed flask stirred with a KPG stirrer; the degree of turnover was continuously determined by HPLC analysis. After a total of 96 h the contents of the reactor were filtered off via a Buchner funnel. The moist filter cake was put back into the round bottom flask and stirred at RT in 300 ml of 50 mM sodium carbonate buffer, pH 10.0 for 5 min, during which part of the isoquercetin dissolved with an intensely yellow color. The suspension was filtered and the filter cake was immediately extracted again with carbonate buffer. After a total of 7 extraction cycles, the eupergite was largely colorless and the isoquercetin was almost completely dissolved. The extracts were combined, carefully acidified with dilute hydrochloric acid until the pH was approximately 3 and then cooled to 4 ° C. After filtration and drying in a desiccant A yield of 4.9 g of product was obtained, which was composed of 98% isoquercetin and 2% rutin.
Das feuchte Eupergit wurde zweimal mit 50 mM Citratpuffer gewaschen und war danach wieder für weitere Umsetzungen einsatzbereit Die Aktivität des Enzyms betrug noch 3,9 U/g, das entspricht einem Aktivitätsverlust von 7%.The moist eupergite was washed twice with 50 mM citrate buffer and was then ready for further reactions. The activity of the enzyme was still 3.9 U / g, which corresponds to a loss of activity of 7%.
Beispiel 4Example 4
1 Modifizierung von magnetischen Silicapartikeln mit Aldehvdgruppen und Immobilisierung von Naringinase auf diesen Partikeln1 Modification of magnetic silica particles with aldehyde groups and immobilization of naringinase on these particles
In einem 1 I Dreihalskolben - ausgestattet mit KPG-Rührer, Tropftrichter und Rückflusskühler - wurde eine Suspension von 30 g magnetischen Silicapartikeln (MagPrep, Merck, Darmstadt) in 600 ml Wasser vorgelegt. Eine Mischung von 20 ml Aminopropyl- triethoxysilan (ABCR, Karlsruhe) und 20 ml Isopropanol wurde innerhalb 30 min unter Rühren zugetropft. Anschließend wurde das Gemisch auf 85°C erhitzt und 1 h lang bei dieser Temperatur gerührt. Nach dem Abkühlen wurde die Suspension in ein Becherglas überführt, die Partikel mittels eines starken Permanentmagneten am Boden des Gefäßes gesammelt und der Überstand dekantiert. Die Partikel wurden wiederholt mit demin. Wasser gewaschen, bis der pH-Wert des Waschwassers konstant blieb. Anschließend wurden die Partikel in 600 ml Wasser resuspendiert und der pH-Wert mit einigen Tropfen Essigsäure auf einen Wert von ca. 8 eingestellt; nach Zugabe von 24 ml 50%iger Glutardialdehydlösung wurde die Suspension 4 h lang bei RT gerührt und die Partikel danach so lange mit demin. Wasser gewaschen, bis im Waschwasser kein Glutaraldehyd mehr nachgewiesen werden konnte (Tüpfelreaktion mit schwefelsaurer 2,4-Dinitrophenylhydrazin-Lösung).A suspension of 30 g magnetic silica particles (MagPrep, Merck, Darmstadt) in 600 ml water was placed in a 1 liter three-necked flask equipped with a KPG stirrer, dropping funnel and reflux condenser. A mixture of 20 ml of aminopropyltriethoxysilane (ABCR, Karlsruhe) and 20 ml of isopropanol was added dropwise with stirring over the course of 30 minutes. The mixture was then heated to 85 ° C. and stirred at this temperature for 1 hour. After cooling, the suspension was transferred to a beaker, the particles were collected at the bottom of the vessel using a strong permanent magnet, and the supernatant was decanted. The particles were repeated with demin. Washed water until the pH of the wash water remained constant. The particles were then resuspended in 600 ml of water and the pH was adjusted to about 8 with a few drops of acetic acid; after the addition of 24 ml of 50% glutardialdehyde solution, the suspension was stirred at RT for 4 h and the particles thereafter with demin. Washed water until no more glutaraldehyde could be detected in the wash water (spot reaction with sulfuric acid 2,4-dinitrophenylhydrazine solution).
Die aldehyd-derivatisierten Partikel wurden in einem 1 I Rundkolben in 600 ml 0,2 M Kaliumphosphatpuffer, pH 9, resuspendiert. Nach Zugabe einer Lösung von 1 g Naringinase (Sigma, Deisenhofen) in 100 ml 50 mM Natriumchloridlösung wurde die Mischung mit einem KPG-Rührer 2 Tage bei RT gerührt. Anschließend wurden die Partikel mit Hilfe eines Permanentmagneten abgetrennt und wiederholt zunächst mit 0,2 M Nat-
riumchloridlösung, dann mit 50 mM Citratpuffer, pH 4,0, gewaschen. Die Rhamnosida- seaktivität des Immobilisats wurde mit p-Nitrophenyl-L- -rhamnopyranosid (Sigma, Dei- senhofen) als Substrat nach der Methode von Kurosawa bestimmt (Kurosawa, Ikeda, Egami, J. Biochem. 73, 31-37 (1973): α-L-Rhamnosidases of the liver of Turbo cornutus and Aspergillus niger); sie betrug 162 U/g.The aldehyde-derivatized particles were resuspended in a 1 liter round-bottom flask in 600 ml of 0.2 M potassium phosphate buffer, pH 9. After adding a solution of 1 g of naringinase (Sigma, Deisenhofen) in 100 ml of 50 mM sodium chloride solution, the mixture was stirred at RT for 2 days using a KPG stirrer. The particles were then separated using a permanent magnet and repeated first with 0.2 M sodium hydroxide. rium chloride solution, then washed with 50 mM citrate buffer, pH 4.0. The rhamnosidase activity of the immobilizate was determined using p-nitrophenyl-L- rhamnopyranoside (Sigma, Deisenhofen) as a substrate using the Kurosawa method (Kurosawa, Ikeda, Egami, J. Biochem. 73, 31-37 (1973) : α-L-Rhamnosidases of the liver of Turbo cornutus and Aspergillus niger); it was 162 U / g.
2. Modifizierung von magnetischen Silicapartikeln mit Epoxyqruppen und Immobilisierung von Naringinase auf diesen Partikeln2. Modification of magnetic silica particles with epoxy groups and immobilization of naringinase on these particles
In einem 1 I Dreihalskolben - ausgestattet mit KPG-Rührer, Tropftrichter und Rückflusskühler - wurde eine Suspension von 30 g magnetischen Silicapartikeln (MagPrep, Merck, Darmstadt) in 600 ml 50 mM Natriumacetatlösung vorgelegt. Eine Mischung von 20 ml (3-Glycidoxypropyl)trimethoxysilan (ABCR, Karlsruhe) und 20 ml Isopropanol wurde innerhalb 30 min unter Rühren zugetropft, anschließend das Gemisch auf 85°C erhitzt und 1 h lang bei dieser Temperatur gerührt. Nach dem Abkühlen wurde die Suspension in ein Becherglas überführt, die Partikel mittels eines starken Permanentmagneten am Boden des Gefäßes gesammelt und der Überstand dekantiert. Die Partikel wurden wiederholt mit demin. Wasser gewaschen, bis der pH-Wert des Waschwassers konstant blieb. Zur quantitativen Bestimmung der Epoxygruppen wurde eine Probe von ca. 0,5 g des Materials wiederholt mit Methanol gewaschen und anschließend bei ca. 70°C im Trockenschrank bis zur Gewichtskonstanz getrocknet. Die Bestimmung der Epoxygruppen nach der Methode von Pribyl (Pribyl, Fresenius Z. Anal. Chem. 303, 113- 116 (1980): Bestimmung von Epoxydendgruppen in modifizierten chromatographischen Sorbentien und Gelen) ergab 250 μmol/g.A suspension of 30 g of magnetic silica particles (MagPrep, Merck, Darmstadt) in 600 ml of 50 mM sodium acetate solution was placed in a 1 liter three-necked flask equipped with a KPG stirrer, dropping funnel and reflux condenser. A mixture of 20 ml (3-glycidoxypropyl) trimethoxysilane (ABCR, Karlsruhe) and 20 ml of isopropanol was added dropwise over the course of 30 minutes with stirring, then the mixture was heated to 85 ° C. and stirred at this temperature for 1 hour. After cooling, the suspension was transferred to a beaker, the particles were collected at the bottom of the vessel using a strong permanent magnet, and the supernatant was decanted. The particles were repeated with demin. Washed water until the pH of the wash water remained constant. For quantitative determination of the epoxy groups, a sample of about 0.5 g of the material was washed repeatedly with methanol and then dried to constant weight at about 70 ° C. in a drying cabinet. The determination of the epoxy groups by the method of Pribyl (Pribyl, Fresenius Z. Anal. Chem. 303, 113-116 (1980): determination of epoxy end groups in modified chromatographic sorbents and gels) gave 250 μmol / g.
150 ml einer 20%igen (w/v) Suspension epoxy-derivatisierter Magnetpartikel wurden in einem 1 I Rundkolben mit 350 ml 1 M Kaliumphosphatpuffer, pH 9,0, vermischt. Nach Zugabe einer Lösung von 1 ,5 g Naringinase (Sigma, Deisenhofen) in 15 ml 50 mM Natriumchloridlösung wurde die Mischung mit einem KPG-Rührer 16 h bei 40°C gerührt. Anschließend wurden die Partikel mit Hilfe eines Permanentmagneten abgetrennt und wiederholt zunächst mit 0,2 M Natriumchloridlösung, dann mit 50 mM Citratpuffer, pH 4,0, gewaschen. Die Rhamnosidaseaktivität des Immobilisats wurde mit p-Nitrophenyl-L-
α-rhamnopyranosid (Sigma, Deisenhofen) als Substrat nach der Methode von Kurosawa bestimmt; sie betrug 102 U/g.150 ml of a 20% (w / v) suspension of epoxy-derivatized magnetic particles were mixed in a 1 l round-bottom flask with 350 ml of 1 M potassium phosphate buffer, pH 9.0. After adding a solution of 1.5 g of naringinase (Sigma, Deisenhofen) in 15 ml of 50 mM sodium chloride solution, the mixture was stirred at 40 ° C. for 16 h using a KPG stirrer. The particles were then separated using a permanent magnet and repeatedly washed first with 0.2 M sodium chloride solution, then with 50 mM citrate buffer, pH 4.0. The rhamnosidase activity of the immobilizate was determined using p-nitrophenyl-L- α-rhamnopyranoside (Sigma, Deisenhofen) determined as substrate by the Kurosawa method; it was 102 U / g.
3. Modifizierung von magnetischen Silicapartikeln mit Carboxylgruppen und Immobilisierung von Naringinase auf diesen Partikeln3. Modification of magnetic silica particles with carboxyl groups and immobilization of naringinase on these particles
In einem 1 I Dreihalskolben - ausgestattet mit KPG-Rührer, Tropftrichter und Rückflusskühler - wurde eine Suspension von 30 g magnetischen Silicapartikeln (MagPrep, Merck, Darmstadt) in 600 ml Wasser vorgelegt. Eine Mischung von 28 ml 3-(Triethoxy- silyl)propylsuccinylanhydrid (ABCR, Karlsruhe) und 28 ml Isopropanol wurde innerhalb 30 min unter Rühren zugetropft und anschließend der pH-Wert der Reaktionsmischung durch tropfenweise Zugabe einer 10%igen Natronlauge auf 9,0 eingestellt. Das Gemisch wurde auf 80°C erhitzt und 2 h lang bei dieser Temperatur gerührt. Dabei wurde in regelmäßigen Abständen der pH kontrolliert und ggf. durch Zugabe von Alkali korrigiert. Nach dem Abkühlen wurde die Suspension in ein Becherglas überführt, die Partikel mittels eines starken Permanentmagneten am Boden des Gefäßes gesammelt und der Überstand dekantiert. Die Partikel wurden dreimal mit demin. Wasser, einmal mit einer 2 M Essigsäurelösung und dann wiederholt so lange mit demin. Wasser gewaschen, bis der pH-Wert des Waschwassers konstant blieb.A suspension of 30 g magnetic silica particles (MagPrep, Merck, Darmstadt) in 600 ml water was placed in a 1 liter three-necked flask equipped with a KPG stirrer, dropping funnel and reflux condenser. A mixture of 28 ml of 3- (triethoxysilyl) propylsuccinyl anhydride (ABCR, Karlsruhe) and 28 ml of isopropanol was added dropwise with stirring over the course of 30 minutes and then the pH of the reaction mixture was adjusted to 9.0 by dropwise addition of a 10% sodium hydroxide solution , The mixture was heated to 80 ° C and stirred at this temperature for 2 hours. The pH was checked at regular intervals and corrected if necessary by adding alkali. After cooling, the suspension was transferred to a beaker, the particles were collected at the bottom of the vessel using a strong permanent magnet, and the supernatant was decanted. The particles were demineralized three times. Water, once with a 2 M acetic acid solution and then repeatedly with demin. Washed water until the pH of the wash water remained constant.
150 ml einer 20%igen (w/v) Suspension carboxyl-derivatisierter Magnetpartikel wurden in einem 1 I Rundkolben mit 300 ml 0,4 M Kaliumphosphatpuffer, pH 5,0, und einer Lösung von 1 ,5 g Naringinase (Sigma, Deisenhofen) in 150 ml 50 mM Natriumchloridlösung vermischt. Nach Zugabe von 8 ml einer 1 %igen (w/v) Lösung von EDC (N-Ethyl- N'-(3-dimethylaminopropyl)-carbodiimidhydrochlorid, Merck, Darmstadt) in Wasser wurde das Gemisch 20 h lang bei RT gerührt. Anschließend wurden die Partikel mit Hilfe eines Permanentmagneten abgetrennt und wiederholt zunächst mit 0,2 M Natriumchloridlösung, dann mit 50 mM Citratpuffer, pH 4,0, gewaschen. Die Rhamnosidase- aktivität des Immobilisats wurde mit p-Nitrophenyl-L-α-rhamnopyranosid (Sigma, Deisenhofen) als Substrat nach der Methode von Kurosawa bestimmt; sie betrug 71 U/g.
4. Modifizierung von magnetischen Glimmerpigmenten mit Aldehvdgruppen und Immobilisierung von Hesperidinase auf diesen Partikeln150 ml of a 20% (w / v) suspension of carboxyl-derivatized magnetic particles were in a 1 l round-bottom flask with 300 ml of 0.4 M potassium phosphate buffer, pH 5.0, and a solution of 1.5 g of naringinase (Sigma, Deisenhofen) mixed in 150 ml of 50 mM sodium chloride solution. After adding 8 ml of a 1% (w / v) solution of EDC (N-ethyl-N '- (3-dimethylaminopropyl) carbodiimide hydrochloride, Merck, Darmstadt) in water, the mixture was stirred at RT for 20 h. The particles were then separated using a permanent magnet and repeatedly washed first with 0.2 M sodium chloride solution, then with 50 mM citrate buffer, pH 4.0. The rhamnosidase activity of the immobilizate was determined using p-nitrophenyl-L-α-rhamnopyranoside (Sigma, Deisenhofen) as substrate by the Kurosawa method; it was 71 U / g. 4. Modification of magnetic mica pigments with aldehyde groups and immobilization of hesperidinase on these particles
In einem 1-l-Dreihalskolben - ausgestattet mit KPG-Rührer, Tropftrichter und Rückflusskühler - wurde eine Suspension von 30 g magnetischen Glimmerpigmenten ("Colo- rona Blackstar Green", Merck, Darmstadt) in 300 ml Wasser vorgelegt. Eine Mischung von 20 ml Aminopropyltriethoxysilan (ABCR, Karlsruhe) und 20 ml Isopropanol wurde innerhalb 30 min unter Rühren zugetropft. Anschließend wurde das Gemisch auf 85°C erhitzt und 1 h lang bei dieser Temperatur gerührt. Nach dem Abkühlen wurde die Suspension in ein Becherglas überführt, die Partikel mittels eines Permanentmagneten am Boden des Gefäßes gesammelt und der Überstand dekantiert. Die Partikel wiederholt mit demin. Wasser gewaschen, bis der pH-Wert des Waschwassers konstant blieb. Anschließend wurden die Partikel in 300 ml Wasser resuspendiert und der pH-Wert mit einigen Tropfen Essigsäure auf einen Wert von ca. 8 eingestellt; nach Zugabe von 25 ml 50%iger Glutardialdehydlösung wurde die Suspension 4 h lang bei RT gerührt und die Partikel danach so lange mit demin. Wasser gewaschen, bis im Waschwasser kein Glutaraldehyd mehr nachgewiesen werden konnte (Tüpfelreaktion mit schwefelsaurer 2,4-Dinitrophenylhydrazin-Lösung).A suspension of 30 g of magnetic mica pigments ("Colorado Blackstar Green", Merck, Darmstadt) in 300 ml of water was placed in a 1 liter three-necked flask equipped with a KPG stirrer, dropping funnel and reflux condenser. A mixture of 20 ml of aminopropyltriethoxysilane (ABCR, Karlsruhe) and 20 ml of isopropanol was added dropwise with stirring over the course of 30 minutes. The mixture was then heated to 85 ° C. and stirred at this temperature for 1 hour. After cooling, the suspension was transferred to a beaker, the particles were collected at the bottom of the vessel using a permanent magnet, and the supernatant was decanted. The particles repeated with demin. Washed water until the pH of the wash water remained constant. The particles were then resuspended in 300 ml of water and the pH was adjusted to about 8 with a few drops of acetic acid; after addition of 25 ml of 50% glutardialdehyde solution, the suspension was stirred for 4 hours at RT and the particles thereafter with demin. Washed water until no more glutaraldehyde could be detected in the wash water (spot reaction with sulfuric acid 2,4-dinitrophenylhydrazine solution).
30 g aldehyd-derivatisierten Glimmerpigmente "Colorona Blackstar Green" wurden in einem 1 I Rundkolben in 300 ml 0,2 M Kaliumphosphatpuffer, pH 7,5, resuspendiert. Nach Zugabe einer Lösung von 1 g Hesperidinase (Amano) in 20 ml 0,2 M Kaliumphosphatpuffer, pH 7,5, wurde die Mischung mit einem KPG-Rührer 3 Tage bei RT gerührt. Anschließend wurden die Partikel mit Hilfe eines Permanentmagneten abgetrennt und wiederholt zunächst mit 0,2 M Natriumchloridlösung, dann mit 50 mM Citratpuffer, pH 4,0, gewaschen. Die Rhamnosidaseaktivität des Immobilisats wurde mit p- Nitrophenyl-L-α-rhamnopyranosid (Sigma, Deisenhofen) als Substrat nach der Methode von Kurosawa bestimmt; sie betrug 10 U/g.
5. Umsetzung von Rutin zu Isoguercetin mit immobilisierter Naringinase im Rührkesselreaktor und Abtrennung des magnetischen Biokatalvsators mit einem Permanentmagneten30 g of aldehyde-derivatized mica pigments "Colorona Blackstar Green" were resuspended in a 1 liter round-bottom flask in 300 ml of 0.2 M potassium phosphate buffer, pH 7.5. After adding a solution of 1 g hesperidinase (Amano) in 20 ml 0.2 M potassium phosphate buffer, pH 7.5, the mixture was stirred with a KPG stirrer for 3 days at RT. The particles were then separated using a permanent magnet and repeatedly washed first with 0.2 M sodium chloride solution, then with 50 mM citrate buffer, pH 4.0. The rhamnosidase activity of the immobilizate was determined using p-nitrophenyl-L-α-rhamnopyranoside (Sigma, Deisenhofen) as substrate by the Kurosawa method; it was 10 U / g. 5. Conversion of rutin to isoguercetin with immobilized naringinase in the stirred tank reactor and separation of the magnetic biocatalyst with a permanent magnet
In einem 500 ml Doppelmantel-Rührreaktor wurden 400 ml 50 mM Citratpuffer, pH 5,0, 20 g auf magnetischen Silicapartikeln immobilisierte Naringinase mit einer Aktivität von 102 U/g und 10 g Rutin (Merck, Darmstadt) bei 40°C miteinander verrührt; der Grad des Umsatzes wurde in Abständen durch HPLC-Analyse bestimmt. Nach 24 h wurde der Reaktorinhalt in ein Becherglas gepumpt und der Katalysator mit Hilfe eines Plattenmagneten (Bakker, 200 mT) am Boden des Gefäßes gesammelt. Der Überstand wurde sofort mit Hilfe einer Pumpe abgesaugt und die Magnetpartikel mehrmals mit jeweils 100 ml Puffer gewaschen, um die letzten Reste an anhaftendem festem Isoquercetin abzuspülen. Das gesammelte Isoquercetin wurde abfiltriert, mehrmals mit geringen Portionen Eiswasser gewaschen und im Exsikkator getrocknet. Die Ausbeute betrug 6,5 g. Eine HPLC-Analyse ergab eine Zusammensetzung von 96% Isoquercetin, 2% Quercetin und 2% Rutin. Die Aktivität des Immobilisats betrug nach der Umsetzung noch 92 U/g. Dies entspricht einem Aktivitätsverlust von 10%.400 ml of 50 mM citrate buffer, pH 5.0, 20 g of naringinase immobilized on magnetic silica particles with an activity of 102 U / g and 10 g of rutin (Merck, Darmstadt) were stirred together at 40 ° C. in a 500 ml double-jacket stirred reactor; the degree of conversion was determined at intervals by HPLC analysis. After 24 hours, the reactor contents were pumped into a beaker and the catalyst was collected at the bottom of the vessel using a plate magnet (Bakker, 200 mT). The supernatant was immediately aspirated using a pump and the magnetic particles were washed several times with 100 ml of buffer in order to rinse off the last residues of adhering solid isoquercetin. The collected isoquercetin was filtered off, washed several times with small portions of ice water and dried in a desiccator. The yield was 6.5 g. HPLC analysis showed a composition of 96% isoquercetin, 2% quercetin and 2% rutin. The activity of the immobilized product was still 92 U / g after the reaction. This corresponds to an activity loss of 10%.
6. Umsetzung von Rutin zu Isoquercetin mit immobilisierter Naringinase im Rührkesselreaktor und Abtrennung des magnetischen Biokatalvsators mit einem elektromagnetischen Separator (Abb.1)6. Conversion of rutin to isoquercetin with immobilized naringinase in a stirred tank reactor and separation of the magnetic biocatalyst with an electromagnetic separator (Fig. 1)
In einem 500 ml Doppelmantel-Rührreaktor wurden 300 ml 50 mM Citratpuffer, pH 5,0, 10 g auf magnetischen Silicapartikeln immobilisierte Naringinase mit einer Aktivität von 102 U/g und 5 g Rutin (Merck, Darmstadt) bei 40°C miteinander verrührt; der Grad des Umsatzes wurde laufend durch HPLC-Analyse bestimmt. Nach 24 h wurde der Reaktorinhalt mit Hilfe einer peristaltischen Pumpe mit einer Flussrate von 25 ml/min durch eine elektromagnetische HGMS-Anlage geleitet, wodurch die Magnetpartikel vollständig an der Drahtmatrix abgeschieden wurden (Technische Daten der Abscheideanlage: Glasrohr mit Innendurchmesser 20 mm und 200 mm Länge, Leervolumen 65 ml, Gewicht der Drahtpackung aus SS-Legierung 15 g, 4 Spulen in Serie geschaltet, Strom-
stärke 6 A, magnet. Feldstärke des Helmholtzfeldes 25 mT). Anschließend wurden zweimal jeweils 100 ml Citratpuffer bei eingeschaltetem Magnetfeld durch die Säule gepumpt, um die Magnetpartikel zu spülen. Die vereinigten Produktsuspensionen wurden filtriert, das Isoquercetin mit Eiswasser gewaschen und im Exsikkator getrocknet. Die Ausbeute betrug 3,1 g. Eine HPLC-Analyse ergab eine Zusammensetzung von 97% Isoquercetin, 2% Quercetin und 1% Rutin.300 ml of 50 mM citrate buffer, pH 5.0, 10 g of naringinase immobilized on magnetic silica particles with an activity of 102 U / g and 5 g of rutin (Merck, Darmstadt) were stirred together at 40 ° C. in a 500 ml double-jacket stirred reactor. the degree of turnover was continuously determined by HPLC analysis. After 24 hours, the reactor contents were passed through an electromagnetic HGMS system using a peristaltic pump with a flow rate of 25 ml / min, whereby the magnetic particles were completely separated on the wire matrix (technical data of the separation system: glass tube with an inner diameter of 20 mm and 200 mm Length, empty volume 65 ml, weight of the wire packing made of SS alloy 15 g, 4 coils connected in series, current 6 A, magnet. Field strength of the Helmholtz field 25 mT). Subsequently, 100 ml of citrate buffer were pumped through the column with the magnetic field switched on in order to rinse the magnetic particles. The combined product suspensions were filtered, the isoquercetin washed with ice water and dried in a desiccator. The yield was 3.1 g. HPLC analysis showed a composition of 97% isoquercetin, 2% quercetin and 1% rutin.
Zur Rückgewinnung des Katalysators wurden 10 min lang bei abgeschaltetem Magnetfeld 100 ml Citratpuffer, pH 5,0, mit einer Durchflussrate von 100 ml/min im Kreislauf durch die Abscheideanlage gepumpt, wobei die Flussrichtung mehrmals gewechselt wurde. Anschließend wurde die Katalysatorsuspension zurück in den Rührkessel gepumpt und die im Abscheider verbliebene restliche Menge an Katalysator erneut zweimal mit jeweils 100 ml Citratpuffer abgespült. Die Aktivität des Immobilisats betrug nach der Umsetzung noch 94 U/g. Dies entspricht einem Aktivitätsverlust von 8%.To recover the catalyst, 100 ml of citrate buffer, pH 5.0, were pumped through the separator at a flow rate of 100 ml / min for 10 minutes with the magnetic field switched off, the direction of flow being changed several times. The catalyst suspension was then pumped back into the stirred tank and the remaining amount of catalyst remaining in the separator was rinsed again twice with 100 ml of citrate buffer. The activity of the immobilizate was still 94 U / g after the reaction. This corresponds to an activity loss of 8%.
7. Umsetzung von Rutin zu Isoquercetin mit auf Glimmerpartikeln immobilisierter Hesperidinase im Rührkesselreaktor und Abtrennung des magnetischen Biokatalvsators mit einem Plattenmagneten7. Conversion of rutin to isoquercetin with hesperidinase immobilized on mica particles in a stirred tank reactor and separation of the magnetic biocatalyst using a plate magnet
In einem 500 ml Doppelmantel-Rührreaktor wurden 400 ml 50 mM Citratpuffer, pH 5,0, 30 g auf "Colorona Blackstar" immobilisierte Hesperidinase mit einer Aktivität von 10 U/g und 5 g Rutin (Merck, Darmstadt) bei 40°C miteinander verrührt; der Grad des Umsatzes wurde laufend durch HPLC-Analyse bestimmt. Nach 24 h wurde der Reaktorinhalt in ein Becherglas gepumpt und der Katalysator mit Hilfe eines Plattenmagneten (Bakker, 200 mT) am Boden des Gefäßes gesammelt. Der Überstand wurde sofort mit Hilfe einer Pumpe abgesaugt und die Magnetpartikel mehrmals mit jeweils 100 ml Puffer gewaschen, um die letzten Reste an anhaftendem festem Isoquercetin abzuspülen. Das gesammelte Isoquercetin wurde abfiltriert, mehrmals mit geringen Portionen Eiswasser gewaschen und im Exsikkator getrocknet. Die Ausbeute betrug 3,3 g. Eine HPLC- Analyse ergab eine Zusammensetzung von 96% Isoquercetin, und 4% Rutin.
Die Aktivität des Immobilisats betrug nach der Umsetzung noch 9,7 U/g. Dies entspricht einem Aktivitätsverlust von 3%.400 ml of 50 mM citrate buffer, pH 5.0, 30 g immobilized on "Colorona Blackstar" with an activity of 10 U / g and 5 g of rutin (Merck, Darmstadt) at 40 ° C. were immobilized in a 500 ml double-jacket stirred reactor stirred; the degree of turnover was continuously determined by HPLC analysis. After 24 hours, the reactor contents were pumped into a beaker and the catalyst was collected at the bottom of the vessel using a plate magnet (Bakker, 200 mT). The supernatant was immediately aspirated using a pump and the magnetic particles were washed several times with 100 ml of buffer in order to rinse off the last residues of adhering solid isoquercetin. The collected isoquercetin was filtered off, washed several times with small portions of ice water and dried in a desiccator. The yield was 3.3 g. HPLC analysis showed a composition of 96% isoquercetin and 4% rutin. The activity of the immobilizate was 9.7 U / g after the reaction. This corresponds to a loss of activity of 3%.
8. Umsetzung von Rutin zu Isoguercetin mit auf magnetischen Kieselgelpartikeln immobilisierter Naringinase in einem MSFB-Reaktor8. Conversion of rutin to isoguercetin with naringinase immobilized on magnetic silica gel particles in an MSFB reactor
In einem Vorlagegefäß wurde eine Mischung aus 5 g Rutin, 900 ml 50 mM Citratpuffer, pH 5,0, und 100 ml Methylacetat bei 40°C so lange gerührt, bis eine annähernd homogene Suspension ohne größere Agglomerate entstanden war. Der Reaktor war nicht temperierbar. Das Methylacetat wurde zugesetzt, um die Löslichkeit und die Nachlöserate bei RT zu erhöhen, sowie um die Bildung von Quercetin zu verhindern. Inzwischen wurde eine Suspension von 6 g auf magnetischen Silicapartikeln immobilisierter Naringinase mit einer Aktivität von 162 U/g in 60 ml 50 mM Citratpuffer, pH 5,0, bei ausgeschaltetem Magnetfeld mit Hilfe einer Pumpe in das Rohr des MSFB-Reaktors befördert. Anschließend wurde ein 20 mT starkes Magnetfeld eingestellt und von unten zunächst frischer Citratpuffer mit Hilfe einer exakt dosierbaren Kolbenhubpumpe mit einem Fluss von 5 ml/min eingeleitet, bis sich die Partikel im Magnetfeld stabilisiert hatten. Dann wurde die Rutinsuspension bei RT innerhalb 3,5 h durch den MSFB-Reaktor gepumpt. Aus dem Produktgemisch wurde zunächst das Methylacetat am Rotationsverdampfer entfernt; dann wurde das Produkt abfiltriert, mehrfach mit Eiswasser gewaschen, und im Exsikkator getrocknet. Die Produktausbeute betrug 2,9 g; das Produkt bestand aus 86% Isoquercetin und 14% Rutin.
A mixture of 5 g of rutin, 900 ml of 50 mM citrate buffer, pH 5.0 and 100 ml of methyl acetate was stirred in a storage vessel at 40 ° C. until an approximately homogeneous suspension without larger agglomerates was formed. The reactor could not be heated. The methyl acetate was added to increase the solubility and redissolving rate at RT and to prevent the formation of quercetin. In the meantime, a suspension of 6 g of naringinase immobilized on magnetic silica particles with an activity of 162 U / g in 60 ml of 50 mM citrate buffer, pH 5.0, was conveyed into the tube of the MSFB reactor using a pump with the magnetic field switched off. A 20 mT magnetic field was then set and fresh citrate buffer was initially introduced from below with the aid of a precisely metered piston stroke pump with a flow of 5 ml / min until the particles had stabilized in the magnetic field. The rutin suspension was then pumped through the MSFB reactor at RT within 3.5 h. The methyl acetate was first removed from the product mixture on a rotary evaporator; then the product was filtered off, washed several times with ice water and dried in a desiccator. The product yield was 2.9 g; the product consisted of 86% isoquercetin and 14% rutin.