WO2003096797A2 - Method of increasing plant organ and seed size in a plant - Google Patents
Method of increasing plant organ and seed size in a plant Download PDFInfo
- Publication number
- WO2003096797A2 WO2003096797A2 PCT/US2003/014989 US0314989W WO03096797A2 WO 2003096797 A2 WO2003096797 A2 WO 2003096797A2 US 0314989 W US0314989 W US 0314989W WO 03096797 A2 WO03096797 A2 WO 03096797A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- seq
- plant
- dna
- promoter
- sequence
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 92
- 210000000056 organ Anatomy 0.000 title claims abstract description 35
- 230000001965 increasing effect Effects 0.000 title claims description 23
- 230000009261 transgenic effect Effects 0.000 claims abstract description 48
- 241000196324 Embryophyta Species 0.000 claims description 322
- 108090000623 proteins and genes Proteins 0.000 claims description 191
- 108020004414 DNA Proteins 0.000 claims description 152
- 102000053602 DNA Human genes 0.000 claims description 131
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 107
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 95
- 229920001184 polypeptide Polymers 0.000 claims description 94
- 102000004169 proteins and genes Human genes 0.000 claims description 69
- 230000006870 function Effects 0.000 claims description 29
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 claims description 23
- 240000008042 Zea mays Species 0.000 claims description 16
- 235000002017 Zea mays subsp mays Nutrition 0.000 claims description 16
- 244000068988 Glycine max Species 0.000 claims description 15
- 235000010469 Glycine max Nutrition 0.000 claims description 13
- 240000001980 Cucurbita pepo Species 0.000 claims description 9
- 240000007594 Oryza sativa Species 0.000 claims description 9
- 235000007164 Oryza sativa Nutrition 0.000 claims description 9
- 235000009566 rice Nutrition 0.000 claims description 9
- 230000001131 transforming effect Effects 0.000 claims description 8
- 244000241257 Cucumis melo Species 0.000 claims description 7
- 244000061456 Solanum tuberosum Species 0.000 claims description 7
- 235000002595 Solanum tuberosum Nutrition 0.000 claims description 7
- 235000011299 Brassica oleracea var botrytis Nutrition 0.000 claims description 6
- 240000003259 Brassica oleracea var. botrytis Species 0.000 claims description 6
- 240000006740 Cichorium endivia Species 0.000 claims description 6
- 241001672694 Citrus reticulata Species 0.000 claims description 6
- 235000009854 Cucurbita moschata Nutrition 0.000 claims description 6
- 235000009852 Cucurbita pepo Nutrition 0.000 claims description 6
- 235000007688 Lycopersicon esculentum Nutrition 0.000 claims description 6
- 240000005561 Musa balbisiana Species 0.000 claims description 6
- 235000002637 Nicotiana tabacum Nutrition 0.000 claims description 6
- 240000004713 Pisum sativum Species 0.000 claims description 6
- 235000010582 Pisum sativum Nutrition 0.000 claims description 6
- 240000003768 Solanum lycopersicum Species 0.000 claims description 6
- 244000078534 Vaccinium myrtillus Species 0.000 claims description 6
- 235000003733 chicria Nutrition 0.000 claims description 6
- 235000007319 Avena orientalis Nutrition 0.000 claims description 5
- 235000004977 Brassica sinapistrum Nutrition 0.000 claims description 5
- 235000002767 Daucus carota Nutrition 0.000 claims description 5
- 244000000626 Daucus carota Species 0.000 claims description 5
- 240000005979 Hordeum vulgare Species 0.000 claims description 5
- 235000007340 Hordeum vulgare Nutrition 0.000 claims description 5
- 240000004658 Medicago sativa Species 0.000 claims description 5
- 235000017587 Medicago sativa ssp. sativa Nutrition 0.000 claims description 5
- 235000021307 Triticum Nutrition 0.000 claims description 5
- 244000098338 Triticum aestivum Species 0.000 claims description 5
- 240000007087 Apium graveolens Species 0.000 claims description 4
- 235000015849 Apium graveolens Dulce Group Nutrition 0.000 claims description 4
- 235000010591 Appio Nutrition 0.000 claims description 4
- 235000007558 Avena sp Nutrition 0.000 claims description 4
- 235000014698 Brassica juncea var multisecta Nutrition 0.000 claims description 4
- 235000006008 Brassica napus var napus Nutrition 0.000 claims description 4
- 240000000385 Brassica napus var. napus Species 0.000 claims description 4
- 240000007124 Brassica oleracea Species 0.000 claims description 4
- 235000003899 Brassica oleracea var acephala Nutrition 0.000 claims description 4
- 235000011301 Brassica oleracea var capitata Nutrition 0.000 claims description 4
- 235000001169 Brassica oleracea var oleracea Nutrition 0.000 claims description 4
- 235000006618 Brassica rapa subsp oleifera Nutrition 0.000 claims description 4
- 235000002566 Capsicum Nutrition 0.000 claims description 4
- 235000015510 Cucumis melo subsp melo Nutrition 0.000 claims description 4
- 240000008067 Cucumis sativus Species 0.000 claims description 4
- 235000010799 Cucumis sativus var sativus Nutrition 0.000 claims description 4
- 244000020551 Helianthus annuus Species 0.000 claims description 4
- 235000003222 Helianthus annuus Nutrition 0.000 claims description 4
- 244000088415 Raphanus sativus Species 0.000 claims description 4
- 235000006140 Raphanus sativus var sativus Nutrition 0.000 claims description 4
- 235000009337 Spinacia oleracea Nutrition 0.000 claims description 4
- 244000300264 Spinacia oleracea Species 0.000 claims description 4
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 claims description 4
- 235000005822 corn Nutrition 0.000 claims description 4
- 235000014571 nuts Nutrition 0.000 claims description 4
- 240000004507 Abelmoschus esculentus Species 0.000 claims description 3
- 235000009436 Actinidia deliciosa Nutrition 0.000 claims description 3
- 244000298697 Actinidia deliciosa Species 0.000 claims description 3
- 235000001674 Agaricus brunnescens Nutrition 0.000 claims description 3
- 235000005254 Allium ampeloprasum Nutrition 0.000 claims description 3
- 240000006108 Allium ampeloprasum Species 0.000 claims description 3
- 244000291564 Allium cepa Species 0.000 claims description 3
- 235000002732 Allium cepa var. cepa Nutrition 0.000 claims description 3
- 244000144730 Amygdalus persica Species 0.000 claims description 3
- 244000099147 Ananas comosus Species 0.000 claims description 3
- 235000007119 Ananas comosus Nutrition 0.000 claims description 3
- 235000017060 Arachis glabrata Nutrition 0.000 claims description 3
- 244000105624 Arachis hypogaea Species 0.000 claims description 3
- 235000010777 Arachis hypogaea Nutrition 0.000 claims description 3
- 235000018262 Arachis monticola Nutrition 0.000 claims description 3
- 244000003416 Asparagus officinalis Species 0.000 claims description 3
- 235000005340 Asparagus officinalis Nutrition 0.000 claims description 3
- 235000000832 Ayote Nutrition 0.000 claims description 3
- 235000016068 Berberis vulgaris Nutrition 0.000 claims description 3
- 241000335053 Beta vulgaris Species 0.000 claims description 3
- 241000219310 Beta vulgaris subsp. vulgaris Species 0.000 claims description 3
- 241000167854 Bourreria succulenta Species 0.000 claims description 3
- 235000004221 Brassica oleracea var gemmifera Nutrition 0.000 claims description 3
- 235000017647 Brassica oleracea var italica Nutrition 0.000 claims description 3
- 244000308368 Brassica oleracea var. gemmifera Species 0.000 claims description 3
- 235000004936 Bromus mango Nutrition 0.000 claims description 3
- 235000009467 Carica papaya Nutrition 0.000 claims description 3
- 240000006432 Carica papaya Species 0.000 claims description 3
- 241000701459 Caulimovirus Species 0.000 claims description 3
- 244000298479 Cichorium intybus Species 0.000 claims description 3
- 244000241235 Citrullus lanatus Species 0.000 claims description 3
- 235000012828 Citrullus lanatus var citroides Nutrition 0.000 claims description 3
- 241000207199 Citrus Species 0.000 claims description 3
- 235000008733 Citrus aurantifolia Nutrition 0.000 claims description 3
- 235000005979 Citrus limon Nutrition 0.000 claims description 3
- 244000131522 Citrus pyriformis Species 0.000 claims description 3
- 240000000560 Citrus x paradisi Species 0.000 claims description 3
- 240000007154 Coffea arabica Species 0.000 claims description 3
- 244000018436 Coriandrum sativum Species 0.000 claims description 3
- 229920000742 Cotton Polymers 0.000 claims description 3
- 235000009847 Cucumis melo var cantalupensis Nutrition 0.000 claims description 3
- 235000015001 Cucumis melo var inodorus Nutrition 0.000 claims description 3
- 240000002495 Cucumis melo var. inodorus Species 0.000 claims description 3
- 235000009804 Cucurbita pepo subsp pepo Nutrition 0.000 claims description 3
- 241000219130 Cucurbita pepo subsp. pepo Species 0.000 claims description 3
- 235000003954 Cucurbita pepo var melopepo Nutrition 0.000 claims description 3
- 235000017788 Cydonia oblonga Nutrition 0.000 claims description 3
- 244000019459 Cynara cardunculus Species 0.000 claims description 3
- 235000019106 Cynara scolymus Nutrition 0.000 claims description 3
- 235000011511 Diospyros Nutrition 0.000 claims description 3
- 244000236655 Diospyros kaki Species 0.000 claims description 3
- 235000014466 Douglas bleu Nutrition 0.000 claims description 3
- 244000024675 Eruca sativa Species 0.000 claims description 3
- 235000014755 Eruca sativa Nutrition 0.000 claims description 3
- 244000004281 Eucalyptus maculata Species 0.000 claims description 3
- 240000006927 Foeniculum vulgare Species 0.000 claims description 3
- 235000004204 Foeniculum vulgare Nutrition 0.000 claims description 3
- 235000016623 Fragaria vesca Nutrition 0.000 claims description 3
- 240000009088 Fragaria x ananassa Species 0.000 claims description 3
- 235000011363 Fragaria x ananassa Nutrition 0.000 claims description 3
- 241000219146 Gossypium Species 0.000 claims description 3
- 235000002678 Ipomoea batatas Nutrition 0.000 claims description 3
- 244000017020 Ipomoea batatas Species 0.000 claims description 3
- 235000003228 Lactuca sativa Nutrition 0.000 claims description 3
- 240000008415 Lactuca sativa Species 0.000 claims description 3
- 241000208682 Liquidambar Species 0.000 claims description 3
- 235000006552 Liquidambar styraciflua Nutrition 0.000 claims description 3
- 241000220225 Malus Species 0.000 claims description 3
- 235000011430 Malus pumila Nutrition 0.000 claims description 3
- 235000015103 Malus silvestris Nutrition 0.000 claims description 3
- 235000014826 Mangifera indica Nutrition 0.000 claims description 3
- 240000007228 Mangifera indica Species 0.000 claims description 3
- 240000003183 Manihot esculenta Species 0.000 claims description 3
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 claims description 3
- 235000003805 Musa ABB Group Nutrition 0.000 claims description 3
- 235000018290 Musa x paradisiaca Nutrition 0.000 claims description 3
- 235000001591 Pachyrhizus erosus Nutrition 0.000 claims description 3
- 244000215747 Pachyrhizus erosus Species 0.000 claims description 3
- 235000018669 Pachyrhizus tuberosus Nutrition 0.000 claims description 3
- 239000006002 Pepper Substances 0.000 claims description 3
- 244000025272 Persea americana Species 0.000 claims description 3
- 235000008673 Persea americana Nutrition 0.000 claims description 3
- 244000062780 Petroselinum sativum Species 0.000 claims description 3
- 235000010627 Phaseolus vulgaris Nutrition 0.000 claims description 3
- 244000046052 Phaseolus vulgaris Species 0.000 claims description 3
- 235000008331 Pinus X rigitaeda Nutrition 0.000 claims description 3
- 241000018646 Pinus brutia Species 0.000 claims description 3
- 235000011613 Pinus brutia Nutrition 0.000 claims description 3
- 241001236219 Pinus echinata Species 0.000 claims description 3
- 235000005018 Pinus echinata Nutrition 0.000 claims description 3
- 235000017339 Pinus palustris Nutrition 0.000 claims description 3
- 235000008577 Pinus radiata Nutrition 0.000 claims description 3
- 241000218621 Pinus radiata Species 0.000 claims description 3
- 235000008566 Pinus taeda Nutrition 0.000 claims description 3
- 241000218679 Pinus taeda Species 0.000 claims description 3
- 235000016761 Piper aduncum Nutrition 0.000 claims description 3
- 240000003889 Piper guineense Species 0.000 claims description 3
- 235000017804 Piper guineense Nutrition 0.000 claims description 3
- 235000008184 Piper nigrum Nutrition 0.000 claims description 3
- 235000015266 Plantago major Nutrition 0.000 claims description 3
- 241000219000 Populus Species 0.000 claims description 3
- 235000009827 Prunus armeniaca Nutrition 0.000 claims description 3
- 244000018633 Prunus armeniaca Species 0.000 claims description 3
- 235000006040 Prunus persica var persica Nutrition 0.000 claims description 3
- 240000001416 Pseudotsuga menziesii Species 0.000 claims description 3
- 235000005386 Pseudotsuga menziesii var menziesii Nutrition 0.000 claims description 3
- 244000294611 Punica granatum Species 0.000 claims description 3
- 235000014360 Punica granatum Nutrition 0.000 claims description 3
- 235000014443 Pyrus communis Nutrition 0.000 claims description 3
- 240000001987 Pyrus communis Species 0.000 claims description 3
- 235000017848 Rubus fruticosus Nutrition 0.000 claims description 3
- 240000007651 Rubus glaucus Species 0.000 claims description 3
- 235000011034 Rubus glaucus Nutrition 0.000 claims description 3
- 235000009122 Rubus idaeus Nutrition 0.000 claims description 3
- 240000000111 Saccharum officinarum Species 0.000 claims description 3
- 235000007201 Saccharum officinarum Nutrition 0.000 claims description 3
- 241000209056 Secale Species 0.000 claims description 3
- 235000007238 Secale cereale Nutrition 0.000 claims description 3
- 235000002597 Solanum melongena Nutrition 0.000 claims description 3
- 244000061458 Solanum melongena Species 0.000 claims description 3
- 235000011684 Sorghum saccharatum Nutrition 0.000 claims description 3
- 235000009184 Spondias indica Nutrition 0.000 claims description 3
- 235000021536 Sugar beet Nutrition 0.000 claims description 3
- 244000269722 Thea sinensis Species 0.000 claims description 3
- 235000011941 Tilia x europaea Nutrition 0.000 claims description 3
- 240000006909 Tilia x europaea Species 0.000 claims description 3
- 235000003095 Vaccinium corymbosum Nutrition 0.000 claims description 3
- 235000017537 Vaccinium myrtillus Nutrition 0.000 claims description 3
- 235000009754 Vitis X bourquina Nutrition 0.000 claims description 3
- 235000012333 Vitis X labruscana Nutrition 0.000 claims description 3
- 240000006365 Vitis vinifera Species 0.000 claims description 3
- 235000014787 Vitis vinifera Nutrition 0.000 claims description 3
- FJJCIZWZNKZHII-UHFFFAOYSA-N [4,6-bis(cyanoamino)-1,3,5-triazin-2-yl]cyanamide Chemical compound N#CNC1=NC(NC#N)=NC(NC#N)=N1 FJJCIZWZNKZHII-UHFFFAOYSA-N 0.000 claims description 3
- 235000016520 artichoke thistle Nutrition 0.000 claims description 3
- 235000000183 arugula Nutrition 0.000 claims description 3
- 235000021029 blackberry Nutrition 0.000 claims description 3
- 235000021014 blueberries Nutrition 0.000 claims description 3
- 235000019693 cherries Nutrition 0.000 claims description 3
- 235000020971 citrus fruits Nutrition 0.000 claims description 3
- 235000004879 dioscorea Nutrition 0.000 claims description 3
- 239000004571 lime Substances 0.000 claims description 3
- 235000020232 peanut Nutrition 0.000 claims description 3
- 235000011197 perejil Nutrition 0.000 claims description 3
- 235000015136 pumpkin Nutrition 0.000 claims description 3
- 235000020354 squash Nutrition 0.000 claims description 3
- 235000013616 tea Nutrition 0.000 claims description 3
- 244000062793 Sorghum vulgare Species 0.000 claims 4
- 241000209763 Avena sativa Species 0.000 claims 2
- 235000010643 Leucaena leucocephala Nutrition 0.000 claims 2
- 240000007472 Leucaena leucocephala Species 0.000 claims 2
- 244000061176 Nicotiana tabacum Species 0.000 claims 2
- 235000019713 millet Nutrition 0.000 claims 2
- 150000007523 nucleic acids Chemical group 0.000 description 103
- 102000039446 nucleic acids Human genes 0.000 description 93
- 108020004707 nucleic acids Proteins 0.000 description 93
- 239000002773 nucleotide Substances 0.000 description 83
- 125000003729 nucleotide group Chemical group 0.000 description 83
- 210000004027 cell Anatomy 0.000 description 81
- 235000018102 proteins Nutrition 0.000 description 63
- 235000001014 amino acid Nutrition 0.000 description 48
- 230000014509 gene expression Effects 0.000 description 48
- 210000001519 tissue Anatomy 0.000 description 44
- 125000003275 alpha amino acid group Chemical group 0.000 description 42
- 229920002477 rna polymer Polymers 0.000 description 42
- 102000040430 polynucleotide Human genes 0.000 description 40
- 108091033319 polynucleotide Proteins 0.000 description 40
- 239000002157 polynucleotide Substances 0.000 description 40
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 36
- 229940024606 amino acid Drugs 0.000 description 34
- 150000001413 amino acids Chemical class 0.000 description 34
- 239000013598 vector Substances 0.000 description 34
- 239000002299 complementary DNA Substances 0.000 description 31
- 108091034117 Oligonucleotide Proteins 0.000 description 30
- 108091028043 Nucleic acid sequence Proteins 0.000 description 29
- 238000003752 polymerase chain reaction Methods 0.000 description 28
- 239000013615 primer Substances 0.000 description 27
- 230000009466 transformation Effects 0.000 description 27
- 241000219194 Arabidopsis Species 0.000 description 26
- 239000013612 plasmid Substances 0.000 description 25
- 102000004190 Enzymes Human genes 0.000 description 24
- 108090000790 Enzymes Proteins 0.000 description 24
- 229940088598 enzyme Drugs 0.000 description 24
- 230000012010 growth Effects 0.000 description 23
- 239000000074 antisense oligonucleotide Substances 0.000 description 21
- 238000012230 antisense oligonucleotides Methods 0.000 description 21
- 238000005516 engineering process Methods 0.000 description 21
- 238000006243 chemical reaction Methods 0.000 description 20
- 230000001105 regulatory effect Effects 0.000 description 20
- 230000035772 mutation Effects 0.000 description 19
- 238000013518 transcription Methods 0.000 description 19
- 230000035897 transcription Effects 0.000 description 19
- 239000000758 substrate Substances 0.000 description 18
- 108020004705 Codon Proteins 0.000 description 17
- 108020004511 Recombinant DNA Proteins 0.000 description 17
- 230000000694 effects Effects 0.000 description 17
- 108020004999 messenger RNA Proteins 0.000 description 17
- 230000009758 senescence Effects 0.000 description 17
- 230000002068 genetic effect Effects 0.000 description 16
- 239000000047 product Substances 0.000 description 16
- 230000014616 translation Effects 0.000 description 16
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 15
- 230000003321 amplification Effects 0.000 description 15
- 238000013507 mapping Methods 0.000 description 15
- 238000003199 nucleic acid amplification method Methods 0.000 description 15
- 241000894007 species Species 0.000 description 15
- 238000013519 translation Methods 0.000 description 15
- 238000003556 assay Methods 0.000 description 14
- 239000012634 fragment Substances 0.000 description 14
- 239000000523 sample Substances 0.000 description 14
- 238000012163 sequencing technique Methods 0.000 description 14
- 241000589158 Agrobacterium Species 0.000 description 13
- 238000010367 cloning Methods 0.000 description 13
- 230000000295 complement effect Effects 0.000 description 13
- 239000003550 marker Substances 0.000 description 13
- 108091026890 Coding region Proteins 0.000 description 12
- 235000016383 Zea mays subsp huehuetenangensis Nutrition 0.000 description 12
- 235000009973 maize Nutrition 0.000 description 12
- 230000004048 modification Effects 0.000 description 12
- 238000012986 modification Methods 0.000 description 12
- 108090000994 Catalytic RNA Proteins 0.000 description 11
- 102000053642 Catalytic RNA Human genes 0.000 description 11
- 230000003115 biocidal effect Effects 0.000 description 11
- 108010031100 chloroplast transit peptides Proteins 0.000 description 11
- 238000002955 isolation Methods 0.000 description 11
- 108091092562 ribozyme Proteins 0.000 description 11
- 239000004009 herbicide Substances 0.000 description 10
- 230000001939 inductive effect Effects 0.000 description 10
- 230000008595 infiltration Effects 0.000 description 10
- 238000001764 infiltration Methods 0.000 description 10
- 210000002706 plastid Anatomy 0.000 description 10
- 241000192656 Nostoc Species 0.000 description 9
- 102000004316 Oxidoreductases Human genes 0.000 description 9
- 108090000854 Oxidoreductases Proteins 0.000 description 9
- 230000000692 anti-sense effect Effects 0.000 description 9
- 230000001580 bacterial effect Effects 0.000 description 9
- 238000009396 hybridization Methods 0.000 description 9
- 229930027917 kanamycin Natural products 0.000 description 9
- 229960000318 kanamycin Drugs 0.000 description 9
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 description 9
- 229930182823 kanamycin A Natural products 0.000 description 9
- 101000844721 Homo sapiens Deleted in malignant brain tumors 1 protein Proteins 0.000 description 8
- 101001111714 Homo sapiens RING-box protein 2 Proteins 0.000 description 8
- 101000901226 Homo sapiens S-arrestin Proteins 0.000 description 8
- 101001000628 Rattus norvegicus Peripheral myelin protein 22 Proteins 0.000 description 8
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 8
- 238000007792 addition Methods 0.000 description 8
- 125000000539 amino acid group Chemical group 0.000 description 8
- 238000013461 design Methods 0.000 description 8
- 230000002363 herbicidal effect Effects 0.000 description 8
- 238000004519 manufacturing process Methods 0.000 description 8
- 238000010369 molecular cloning Methods 0.000 description 8
- 230000008488 polyadenylation Effects 0.000 description 8
- 239000000243 solution Substances 0.000 description 8
- 150000003431 steroids Chemical class 0.000 description 8
- 238000006467 substitution reaction Methods 0.000 description 8
- 101100148681 Arabidopsis thaliana SAG13 gene Proteins 0.000 description 7
- 241000588724 Escherichia coli Species 0.000 description 7
- 239000005562 Glyphosate Substances 0.000 description 7
- 238000002835 absorbance Methods 0.000 description 7
- XDDAORKBJWWYJS-UHFFFAOYSA-N glyphosate Chemical compound OC(=O)CNCP(O)(O)=O XDDAORKBJWWYJS-UHFFFAOYSA-N 0.000 description 7
- 229940097068 glyphosate Drugs 0.000 description 7
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 7
- 108010058731 nopaline synthase Proteins 0.000 description 7
- 230000008929 regeneration Effects 0.000 description 7
- 238000011069 regeneration method Methods 0.000 description 7
- 230000001850 reproductive effect Effects 0.000 description 7
- 239000002689 soil Substances 0.000 description 7
- 230000001954 sterilising effect Effects 0.000 description 7
- 108091093088 Amplicon Proteins 0.000 description 6
- 108020005544 Antisense RNA Proteins 0.000 description 6
- 102100031262 Deleted in malignant brain tumors 1 protein Human genes 0.000 description 6
- 108091092195 Intron Proteins 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 210000003763 chloroplast Anatomy 0.000 description 6
- 238000011161 development Methods 0.000 description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- 230000001404 mediated effect Effects 0.000 description 6
- 244000005700 microbiome Species 0.000 description 6
- 230000010076 replication Effects 0.000 description 6
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 6
- 150000003839 salts Chemical class 0.000 description 6
- 238000004659 sterilization and disinfection Methods 0.000 description 6
- 241000589155 Agrobacterium tumefaciens Species 0.000 description 5
- 240000002791 Brassica napus Species 0.000 description 5
- 108020004635 Complementary DNA Proteins 0.000 description 5
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 5
- 108700023224 Glucose-1-phosphate adenylyltransferases Proteins 0.000 description 5
- 108010068370 Glutens Proteins 0.000 description 5
- 101710089395 Oleosin Proteins 0.000 description 5
- 108700008625 Reporter Genes Proteins 0.000 description 5
- 241000700605 Viruses Species 0.000 description 5
- 230000008859 change Effects 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 239000003184 complementary RNA Substances 0.000 description 5
- 230000018109 developmental process Effects 0.000 description 5
- 229910052738 indium Inorganic materials 0.000 description 5
- 230000002018 overexpression Effects 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 238000012545 processing Methods 0.000 description 5
- 210000001938 protoplast Anatomy 0.000 description 5
- 108091008146 restriction endonucleases Proteins 0.000 description 5
- 238000002741 site-directed mutagenesis Methods 0.000 description 5
- 229960000268 spectinomycin Drugs 0.000 description 5
- UNFWWIHTNXNPBV-WXKVUWSESA-N spectinomycin Chemical compound O([C@@H]1[C@@H](NC)[C@@H](O)[C@H]([C@@H]([C@H]1O1)O)NC)[C@]2(O)[C@H]1O[C@H](C)CC2=O UNFWWIHTNXNPBV-WXKVUWSESA-N 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 241000219195 Arabidopsis thaliana Species 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 235000011293 Brassica napus Nutrition 0.000 description 4
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 4
- 239000007836 KH2PO4 Substances 0.000 description 4
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 4
- NWBJYWHLCVSVIJ-UHFFFAOYSA-N N-benzyladenine Chemical compound N=1C=NC=2NC=NC=2C=1NCC1=CC=CC=C1 NWBJYWHLCVSVIJ-UHFFFAOYSA-N 0.000 description 4
- 241000208125 Nicotiana Species 0.000 description 4
- 108700001094 Plant Genes Proteins 0.000 description 4
- 108010039811 Starch synthase Proteins 0.000 description 4
- 108010043934 Sucrose synthase Proteins 0.000 description 4
- 108700019146 Transgenes Proteins 0.000 description 4
- 239000003242 anti bacterial agent Substances 0.000 description 4
- 230000010310 bacterial transformation Effects 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 229960005091 chloramphenicol Drugs 0.000 description 4
- WIIZWVCIJKGZOK-RKDXNWHRSA-N chloramphenicol Chemical compound ClC(Cl)C(=O)N[C@H](CO)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 WIIZWVCIJKGZOK-RKDXNWHRSA-N 0.000 description 4
- 210000000349 chromosome Anatomy 0.000 description 4
- 238000010276 construction Methods 0.000 description 4
- 230000001419 dependent effect Effects 0.000 description 4
- 238000003306 harvesting Methods 0.000 description 4
- 230000006872 improvement Effects 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 229910052751 metal Inorganic materials 0.000 description 4
- 239000002184 metal Substances 0.000 description 4
- 150000002739 metals Chemical class 0.000 description 4
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 4
- 210000003463 organelle Anatomy 0.000 description 4
- GNSKLFRGEWLPPA-UHFFFAOYSA-M potassium dihydrogen phosphate Chemical compound [K+].OP(O)([O-])=O GNSKLFRGEWLPPA-UHFFFAOYSA-M 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- 238000011144 upstream manufacturing Methods 0.000 description 4
- CBMYJHIOYJEBSB-UHFFFAOYSA-N (10S)-3t.17t-Dihydroxy-10r.13c-dimethyl-(5cH.8cH.9tH.14tH)-hexadecahydro-1H-cyclopenta[a]phenanthren Natural products C1C(O)CCC2(C)C3CCC(C)(C(CC4)O)C4C3CCC21 CBMYJHIOYJEBSB-UHFFFAOYSA-N 0.000 description 3
- RAJWOBJTTGJROA-UHFFFAOYSA-N (5alpha)-androstane-3,17-dione Natural products C1C(=O)CCC2(C)C3CCC(C)(C(CC4)=O)C4C3CCC21 RAJWOBJTTGJROA-UHFFFAOYSA-N 0.000 description 3
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 3
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 3
- UPMXNNIRAGDFEH-UHFFFAOYSA-N 3,5-dibromo-4-hydroxybenzonitrile Chemical compound OC1=C(Br)C=C(C#N)C=C1Br UPMXNNIRAGDFEH-UHFFFAOYSA-N 0.000 description 3
- RAJWOBJTTGJROA-WZNAKSSCSA-N 5alpha-androstane-3,17-dione Chemical compound C1C(=O)CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC[C@H]21 RAJWOBJTTGJROA-WZNAKSSCSA-N 0.000 description 3
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 3
- 239000004475 Arginine Substances 0.000 description 3
- 241001167018 Aroa Species 0.000 description 3
- 244000075850 Avena orientalis Species 0.000 description 3
- 241000894006 Bacteria Species 0.000 description 3
- 239000005489 Bromoxynil Substances 0.000 description 3
- 241000701489 Cauliflower mosaic virus Species 0.000 description 3
- 108700010070 Codon Usage Proteins 0.000 description 3
- 239000003155 DNA primer Substances 0.000 description 3
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 3
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 101100491986 Emericella nidulans (strain FGSC A4 / ATCC 38163 / CBS 112.46 / NRRL 194 / M139) aromA gene Proteins 0.000 description 3
- 241000701484 Figwort mosaic virus Species 0.000 description 3
- 102000053187 Glucuronidase Human genes 0.000 description 3
- 108010060309 Glucuronidase Proteins 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 3
- IAJOBQBIJHVGMQ-UHFFFAOYSA-N Phosphinothricin Natural products CP(O)(=O)CCC(N)C(O)=O IAJOBQBIJHVGMQ-UHFFFAOYSA-N 0.000 description 3
- 108010003581 Ribulose-bisphosphate carboxylase Proteins 0.000 description 3
- 238000002105 Southern blotting Methods 0.000 description 3
- 230000032683 aging Effects 0.000 description 3
- 229940088710 antibiotic agent Drugs 0.000 description 3
- -1 antibodies Proteins 0.000 description 3
- 101150037081 aroA gene Proteins 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 3
- 230000015556 catabolic process Effects 0.000 description 3
- 238000004113 cell culture Methods 0.000 description 3
- 239000013599 cloning vector Substances 0.000 description 3
- 238000011109 contamination Methods 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- 230000037430 deletion Effects 0.000 description 3
- 238000012217 deletion Methods 0.000 description 3
- 239000003623 enhancer Substances 0.000 description 3
- 230000007613 environmental effect Effects 0.000 description 3
- 239000013604 expression vector Substances 0.000 description 3
- 230000035784 germination Effects 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 238000011065 in-situ storage Methods 0.000 description 3
- 230000000977 initiatory effect Effects 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 239000011159 matrix material Substances 0.000 description 3
- 230000000442 meristematic effect Effects 0.000 description 3
- 230000031787 nutrient reservoir activity Effects 0.000 description 3
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 3
- 230000008121 plant development Effects 0.000 description 3
- 230000004481 post-translational protein modification Effects 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 230000001172 regenerating effect Effects 0.000 description 3
- 229960004889 salicylic acid Drugs 0.000 description 3
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 3
- 238000010561 standard procedure Methods 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 230000002103 transcriptional effect Effects 0.000 description 3
- 238000011426 transformation method Methods 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- JLIDBLDQVAYHNE-YKALOCIXSA-N (+)-Abscisic acid Chemical compound OC(=O)/C=C(/C)\C=C\[C@@]1(O)C(C)=CC(=O)CC1(C)C JLIDBLDQVAYHNE-YKALOCIXSA-N 0.000 description 2
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- 101150025032 13 gene Proteins 0.000 description 2
- CAAMSDWKXXPUJR-UHFFFAOYSA-N 3,5-dihydro-4H-imidazol-4-one Chemical compound O=C1CNC=N1 CAAMSDWKXXPUJR-UHFFFAOYSA-N 0.000 description 2
- UMCMPZBLKLEWAF-BCTGSCMUSA-N 3-[(3-cholamidopropyl)dimethylammonio]propane-1-sulfonate Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCC[N+](C)(C)CCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 UMCMPZBLKLEWAF-BCTGSCMUSA-N 0.000 description 2
- 108010020183 3-phosphoshikimate 1-carboxyvinyltransferase Proteins 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- QQXLDOJGLXJCSE-UHFFFAOYSA-N 8-methyl-8-azabicyclo[3.2.1]octan-3-one Chemical compound C1C(=O)CC2CCC1N2C QQXLDOJGLXJCSE-UHFFFAOYSA-N 0.000 description 2
- 108010000700 Acetolactate synthase Proteins 0.000 description 2
- 108010085238 Actins Proteins 0.000 description 2
- 108700028369 Alleles Proteins 0.000 description 2
- 101100288144 Arabidopsis thaliana KNAT1 gene Proteins 0.000 description 2
- 229930192334 Auxin Natural products 0.000 description 2
- 101100507655 Canis lupus familiaris HSPA1 gene Proteins 0.000 description 2
- 102000012410 DNA Ligases Human genes 0.000 description 2
- 108010061982 DNA Ligases Proteins 0.000 description 2
- 239000003298 DNA probe Substances 0.000 description 2
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 2
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 2
- 108010054576 Deoxyribonuclease EcoRI Proteins 0.000 description 2
- 108090000204 Dipeptidase 1 Proteins 0.000 description 2
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 2
- 101100437498 Escherichia coli (strain K12) uidA gene Proteins 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- 108010074122 Ferredoxins Proteins 0.000 description 2
- 239000005561 Glufosinate Substances 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 2
- 108060001084 Luciferase Proteins 0.000 description 2
- XUMBMVFBXHLACL-UHFFFAOYSA-N Melanin Chemical compound O=C1C(=O)C(C2=CNC3=C(C(C(=O)C4=C32)=O)C)=C2C4=CNC2=C1C XUMBMVFBXHLACL-UHFFFAOYSA-N 0.000 description 2
- 108020004711 Nucleic Acid Probes Proteins 0.000 description 2
- 108020005187 Oligonucleotide Probes Proteins 0.000 description 2
- 108700026244 Open Reading Frames Proteins 0.000 description 2
- 238000012408 PCR amplification Methods 0.000 description 2
- 238000002123 RNA extraction Methods 0.000 description 2
- 108091027981 Response element Proteins 0.000 description 2
- 108020005543 Satellite RNA Proteins 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- 108020004566 Transfer RNA Proteins 0.000 description 2
- 108010055615 Zein Proteins 0.000 description 2
- 229920002494 Zein Polymers 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- DZBUGLKDJFMEHC-UHFFFAOYSA-N acridine Chemical compound C1=CC=CC2=CC3=CC=CC=C3N=C21 DZBUGLKDJFMEHC-UHFFFAOYSA-N 0.000 description 2
- 108090000637 alpha-Amylases Proteins 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 239000002363 auxin Substances 0.000 description 2
- 101150103518 bar gene Proteins 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- GINJFDRNADDBIN-FXQIFTODSA-N bilanafos Chemical compound OC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](N)CCP(C)(O)=O GINJFDRNADDBIN-FXQIFTODSA-N 0.000 description 2
- 239000003139 biocide Substances 0.000 description 2
- 230000031018 biological processes and functions Effects 0.000 description 2
- 210000004899 c-terminal region Anatomy 0.000 description 2
- 239000003593 chromogenic compound Substances 0.000 description 2
- 239000013611 chromosomal DNA Substances 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 239000000356 contaminant Substances 0.000 description 2
- 230000005014 ectopic expression Effects 0.000 description 2
- 235000013399 edible fruits Nutrition 0.000 description 2
- 238000004520 electroporation Methods 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 230000037433 frameshift Effects 0.000 description 2
- 230000004345 fruit ripening Effects 0.000 description 2
- 230000004927 fusion Effects 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 238000001502 gel electrophoresis Methods 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- 230000008676 import Effects 0.000 description 2
- 238000007901 in situ hybridization Methods 0.000 description 2
- SEOVTRFCIGRIMH-UHFFFAOYSA-N indole-3-acetic acid Chemical compound C1=CC=C2C(CC(=O)O)=CNC2=C1 SEOVTRFCIGRIMH-UHFFFAOYSA-N 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- 229960000310 isoleucine Drugs 0.000 description 2
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 2
- 238000002372 labelling Methods 0.000 description 2
- 230000014634 leaf senescence Effects 0.000 description 2
- 229910001629 magnesium chloride Inorganic materials 0.000 description 2
- 210000001161 mammalian embryo Anatomy 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 239000000203 mixture Substances 0.000 description 2
- 238000002703 mutagenesis Methods 0.000 description 2
- 231100000350 mutagenesis Toxicity 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000002853 nucleic acid probe Substances 0.000 description 2
- 210000004940 nucleus Anatomy 0.000 description 2
- 239000002751 oligonucleotide probe Substances 0.000 description 2
- 230000001590 oxidative effect Effects 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 238000011176 pooling Methods 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- ZCCUUQDIBDJBTK-UHFFFAOYSA-N psoralen Chemical compound C1=C2OC(=O)C=CC2=CC2=C1OC=C2 ZCCUUQDIBDJBTK-UHFFFAOYSA-N 0.000 description 2
- 239000012264 purified product Substances 0.000 description 2
- 239000011535 reaction buffer Substances 0.000 description 2
- 108020004418 ribosomal RNA Proteins 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 238000002864 sequence alignment Methods 0.000 description 2
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 2
- 239000011550 stock solution Substances 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 2
- 239000004474 valine Substances 0.000 description 2
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- BEJKOYIMCGMNRB-GRHHLOCNSA-N (2s)-2-amino-3-(4-hydroxyphenyl)propanoic acid;(2s)-2-amino-3-phenylpropanoic acid Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1.OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 BEJKOYIMCGMNRB-GRHHLOCNSA-N 0.000 description 1
- PSLCKQYQNVNTQI-BHFSHLQUSA-N (2s)-2-aminobutanedioic acid;(2s)-2-aminopentanedioic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O.OC(=O)[C@@H](N)CCC(O)=O PSLCKQYQNVNTQI-BHFSHLQUSA-N 0.000 description 1
- FCHBECOAGZMTFE-ZEQKJWHPSA-N (6r,7r)-3-[[2-[[4-(dimethylamino)phenyl]diazenyl]pyridin-1-ium-1-yl]methyl]-8-oxo-7-[(2-thiophen-2-ylacetyl)amino]-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate Chemical compound C1=CC(N(C)C)=CC=C1N=NC1=CC=CC=[N+]1CC1=C(C([O-])=O)N2C(=O)[C@@H](NC(=O)CC=3SC=CC=3)[C@H]2SC1 FCHBECOAGZMTFE-ZEQKJWHPSA-N 0.000 description 1
- UKAUYVFTDYCKQA-UHFFFAOYSA-N -2-Amino-4-hydroxybutanoic acid Natural products OC(=O)C(N)CCO UKAUYVFTDYCKQA-UHFFFAOYSA-N 0.000 description 1
- 150000005206 1,2-dihydroxybenzenes Chemical class 0.000 description 1
- 101710140048 2S seed storage protein Proteins 0.000 description 1
- 102100029077 3-hydroxy-3-methylglutaryl-coenzyme A reductase Human genes 0.000 description 1
- 101710158485 3-hydroxy-3-methylglutaryl-coenzyme A reductase Proteins 0.000 description 1
- VXGRJERITKFWPL-UHFFFAOYSA-N 4',5'-Dihydropsoralen Natural products C1=C2OC(=O)C=CC2=CC2=C1OCC2 VXGRJERITKFWPL-UHFFFAOYSA-N 0.000 description 1
- HZWWPUTXBJEENE-UHFFFAOYSA-N 5-amino-2-[[1-[5-amino-2-[[1-[2-amino-3-(4-hydroxyphenyl)propanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoic acid Chemical compound C1CCC(C(=O)NC(CCC(N)=O)C(=O)N2C(CCC2)C(=O)NC(CCC(N)=O)C(O)=O)N1C(=O)C(N)CC1=CC=C(O)C=C1 HZWWPUTXBJEENE-UHFFFAOYSA-N 0.000 description 1
- 230000005730 ADP ribosylation Effects 0.000 description 1
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 1
- 101000818108 Acholeplasma phage L2 Uncharacterized 81.3 kDa protein Proteins 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 241000242764 Aequorea victoria Species 0.000 description 1
- 241000589156 Agrobacterium rhizogenes Species 0.000 description 1
- 244000144725 Amygdalus communis Species 0.000 description 1
- 241000208223 Anacardiaceae Species 0.000 description 1
- 102100034613 Annexin A2 Human genes 0.000 description 1
- 108090000668 Annexin A2 Proteins 0.000 description 1
- 102100034612 Annexin A4 Human genes 0.000 description 1
- 108090000669 Annexin A4 Proteins 0.000 description 1
- 241000208173 Apiaceae Species 0.000 description 1
- 108700007757 Arabidopsis GSTF8 Proteins 0.000 description 1
- 108010037365 Arabidopsis Proteins Proteins 0.000 description 1
- 101100059544 Arabidopsis thaliana CDC5 gene Proteins 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- 241000726301 Avocado sunblotch viroid Species 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- 101150077012 BEL1 gene Proteins 0.000 description 1
- 101710084635 Basic endochitinase Proteins 0.000 description 1
- KHBQMWCZKVMBLN-UHFFFAOYSA-N Benzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1 KHBQMWCZKVMBLN-UHFFFAOYSA-N 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 241000219193 Brassicaceae Species 0.000 description 1
- 108010000755 Bromoxynil nitrilase Proteins 0.000 description 1
- QCMYYKRYFNMIEC-UHFFFAOYSA-N COP(O)=O Chemical class COP(O)=O QCMYYKRYFNMIEC-UHFFFAOYSA-N 0.000 description 1
- 235000009025 Carya illinoensis Nutrition 0.000 description 1
- 244000068645 Carya illinoensis Species 0.000 description 1
- 229930186147 Cephalosporin Natural products 0.000 description 1
- 108010022172 Chitinases Proteins 0.000 description 1
- 108010035563 Chloramphenicol O-acetyltransferase Proteins 0.000 description 1
- 108010007108 Chloroplast Thioredoxins Proteins 0.000 description 1
- 108020004638 Circular DNA Proteins 0.000 description 1
- 108091028075 Circular RNA Proteins 0.000 description 1
- 108091035707 Consensus sequence Proteins 0.000 description 1
- 241000219104 Cucurbitaceae Species 0.000 description 1
- IMXSCCDUAFEIOE-UHFFFAOYSA-N D-Octopin Natural products OC(=O)C(C)NC(C(O)=O)CCCN=C(N)N IMXSCCDUAFEIOE-UHFFFAOYSA-N 0.000 description 1
- LMKYZBGVKHTLTN-NKWVEPMBSA-N D-nopaline Chemical compound NC(=N)NCCC[C@@H](C(O)=O)N[C@@H](C(O)=O)CCC(O)=O LMKYZBGVKHTLTN-NKWVEPMBSA-N 0.000 description 1
- IMXSCCDUAFEIOE-RITPCOANSA-N D-octopine Chemical compound [O-]C(=O)[C@@H](C)[NH2+][C@H](C([O-])=O)CCCNC(N)=[NH2+] IMXSCCDUAFEIOE-RITPCOANSA-N 0.000 description 1
- 101150074155 DHFR gene Proteins 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 240000008853 Datura stramonium Species 0.000 description 1
- 102000016680 Dioxygenases Human genes 0.000 description 1
- 108010028143 Dioxygenases Proteins 0.000 description 1
- AHMIDUVKSGCHAU-UHFFFAOYSA-N Dopaquinone Natural products OC(=O)C(N)CC1=CC(=O)C(=O)C=C1 AHMIDUVKSGCHAU-UHFFFAOYSA-N 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- PLUBXMRUUVWRLT-UHFFFAOYSA-N Ethyl methanesulfonate Chemical compound CCOS(C)(=O)=O PLUBXMRUUVWRLT-UHFFFAOYSA-N 0.000 description 1
- 241000206602 Eukaryota Species 0.000 description 1
- 108700024394 Exon Proteins 0.000 description 1
- 101150020286 FIS2 gene Proteins 0.000 description 1
- 241000220485 Fabaceae Species 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 1
- 241000415263 Gibasis Species 0.000 description 1
- 108010061711 Gliadin Proteins 0.000 description 1
- 108010044091 Globulins Proteins 0.000 description 1
- 102000006395 Globulins Human genes 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 1
- 102000004144 Green Fluorescent Proteins Human genes 0.000 description 1
- 101150056327 HMG2 gene Proteins 0.000 description 1
- 101000912350 Haemophilus phage HP1 (strain HP1c1) DNA N-6-adenine-methyltransferase Proteins 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 108091006054 His-tagged proteins Proteins 0.000 description 1
- 101000798222 Homo sapiens Antizyme inhibitor 2 Proteins 0.000 description 1
- 101000827338 Homo sapiens Mitochondrial fission 1 protein Proteins 0.000 description 1
- 206010020649 Hyperkeratosis Diseases 0.000 description 1
- 241000758791 Juglandaceae Species 0.000 description 1
- 101000790844 Klebsiella pneumoniae Uncharacterized 24.8 kDa protein in cps region Proteins 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- WTDRDQBEARUVNC-UHFFFAOYSA-N L-Dopa Natural products OC(=O)C(N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-UHFFFAOYSA-N 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- AHMIDUVKSGCHAU-LURJTMIESA-N L-dopaquinone Chemical compound [O-]C(=O)[C@@H]([NH3+])CC1=CC(=O)C(=O)C=C1 AHMIDUVKSGCHAU-LURJTMIESA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- FFFHZYDWPBMWHY-VKHMYHEASA-N L-homocysteine Chemical compound OC(=O)[C@@H](N)CCS FFFHZYDWPBMWHY-VKHMYHEASA-N 0.000 description 1
- UKAUYVFTDYCKQA-VKHMYHEASA-N L-homoserine Chemical compound OC(=O)[C@@H](N)CCO UKAUYVFTDYCKQA-VKHMYHEASA-N 0.000 description 1
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- LRQKBLKVPFOOQJ-YFKPBYRVSA-N L-norleucine Chemical compound CCCC[C@H]([NH3+])C([O-])=O LRQKBLKVPFOOQJ-YFKPBYRVSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 1
- 101000839464 Leishmania braziliensis Heat shock 70 kDa protein Proteins 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 108010034715 Light-Harvesting Protein Complexes Proteins 0.000 description 1
- 241000209510 Liliopsida Species 0.000 description 1
- 241000724705 Lucerne transient streak virus Species 0.000 description 1
- 239000006142 Luria-Bertani Agar Substances 0.000 description 1
- 239000006137 Luria-Bertani broth Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 101150115300 MAC1 gene Proteins 0.000 description 1
- 240000009200 Macaranga tanarius Species 0.000 description 1
- 235000000487 Macaranga tanarius Nutrition 0.000 description 1
- 108091027974 Mature messenger RNA Proteins 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 102100023845 Mitochondrial fission 1 protein Human genes 0.000 description 1
- 108010085220 Multiprotein Complexes Proteins 0.000 description 1
- 102000007474 Multiprotein Complexes Human genes 0.000 description 1
- FUSGACRLAFQQRL-UHFFFAOYSA-N N-Ethyl-N-nitrosourea Chemical compound CCN(N=O)C(N)=O FUSGACRLAFQQRL-UHFFFAOYSA-N 0.000 description 1
- PYUSHNKNPOHWEZ-YFKPBYRVSA-N N-formyl-L-methionine Chemical compound CSCC[C@@H](C(O)=O)NC=O PYUSHNKNPOHWEZ-YFKPBYRVSA-N 0.000 description 1
- 125000000729 N-terminal amino-acid group Chemical group 0.000 description 1
- 101710202365 Napin Proteins 0.000 description 1
- 241001045988 Neogene Species 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 101000598243 Nicotiana tabacum Probable aquaporin TIP-type RB7-18C Proteins 0.000 description 1
- 101000655028 Nicotiana tabacum Probable aquaporin TIP-type RB7-5A Proteins 0.000 description 1
- 108010033272 Nitrilase Proteins 0.000 description 1
- 108010025915 Nitrite Reductases Proteins 0.000 description 1
- 108091092724 Noncoding DNA Proteins 0.000 description 1
- 241000424623 Nostoc punctiforme Species 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 240000004370 Pastinaca sativa Species 0.000 description 1
- 235000017769 Pastinaca sativa subsp sativa Nutrition 0.000 description 1
- 101710091688 Patatin Proteins 0.000 description 1
- 108010068204 Peptide Elongation Factors Proteins 0.000 description 1
- 240000007377 Petunia x hybrida Species 0.000 description 1
- 241000254064 Photinus pyralis Species 0.000 description 1
- 241000758706 Piperaceae Species 0.000 description 1
- 241000209504 Poaceae Species 0.000 description 1
- 108010076504 Protein Sorting Signals Proteins 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 108020004518 RNA Probes Proteins 0.000 description 1
- 239000003391 RNA probe Substances 0.000 description 1
- 230000004570 RNA-binding Effects 0.000 description 1
- 108091081062 Repeated sequence (DNA) Proteins 0.000 description 1
- 241000612182 Rexea solandri Species 0.000 description 1
- 241000701507 Rice tungro bacilliform virus Species 0.000 description 1
- 101150012041 SK2 gene Proteins 0.000 description 1
- 241000608282 Sagiyama virus Species 0.000 description 1
- 241000242583 Scyphozoa Species 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 101150019148 Slc7a3 gene Proteins 0.000 description 1
- 241000208292 Solanaceae Species 0.000 description 1
- 241000724704 Solanum nodiflorum mottle virus Species 0.000 description 1
- 240000003829 Sorghum propinquum Species 0.000 description 1
- 108091081024 Start codon Proteins 0.000 description 1
- 229930182558 Sterol Natural products 0.000 description 1
- 241000724703 Subterranean clover mottle virus Species 0.000 description 1
- 241000724803 Sugarcane bacilliform virus Species 0.000 description 1
- 229940100389 Sulfonylurea Drugs 0.000 description 1
- 239000005864 Sulphur Substances 0.000 description 1
- 108020005038 Terminator Codon Proteins 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical class OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- 241000723677 Tobacco ringspot virus Species 0.000 description 1
- 102000004357 Transferases Human genes 0.000 description 1
- 108090000992 Transferases Proteins 0.000 description 1
- 241000219793 Trifolium Species 0.000 description 1
- 108700037590 Tropinone reductases Proteins 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 108060008724 Tyrosinase Proteins 0.000 description 1
- 241001002356 Valeriana edulis Species 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 1
- 241000724701 Velvet tobacco mottle virus Species 0.000 description 1
- 241001464837 Viridiplantae Species 0.000 description 1
- 241000726445 Viroids Species 0.000 description 1
- 101001036768 Zea mays Glucose-1-phosphate adenylyltransferase large subunit 1, chloroplastic/amyloplastic Proteins 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- 101150067314 aadA gene Proteins 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000011543 agarose gel Substances 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 108010050181 aleurone Proteins 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 235000020224 almond Nutrition 0.000 description 1
- 102000004139 alpha-Amylases Human genes 0.000 description 1
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 1
- 102000005840 alpha-Galactosidase Human genes 0.000 description 1
- 108010030291 alpha-Galactosidase Proteins 0.000 description 1
- 229940024171 alpha-amylase Drugs 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229940126575 aminoglycoside Drugs 0.000 description 1
- AEMFNILZOJDQLW-QAGGRKNESA-N androst-4-ene-3,17-dione Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1 AEMFNILZOJDQLW-QAGGRKNESA-N 0.000 description 1
- 229960005471 androstenedione Drugs 0.000 description 1
- AEMFNILZOJDQLW-UHFFFAOYSA-N androstenedione Natural products O=C1CCC2(C)C3CCC(C)(C(CC4)=O)C4C3CCC2=C1 AEMFNILZOJDQLW-UHFFFAOYSA-N 0.000 description 1
- 229930002877 anthocyanin Natural products 0.000 description 1
- 235000010208 anthocyanin Nutrition 0.000 description 1
- 239000004410 anthocyanin Substances 0.000 description 1
- 150000004636 anthocyanins Chemical class 0.000 description 1
- 230000009833 antibody interaction Effects 0.000 description 1
- 229940019748 antifibrinolytic proteinase inhibitors Drugs 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 238000002869 basic local alignment search tool Methods 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 102000006635 beta-lactamase Human genes 0.000 description 1
- 230000027455 binding Effects 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 239000007844 bleaching agent Substances 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- 239000012152 bradford reagent Substances 0.000 description 1
- 150000001647 brassinosteroids Chemical class 0.000 description 1
- 238000010804 cDNA synthesis Methods 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- FPPNZSSZRUTDAP-UWFZAAFLSA-N carbenicillin Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)C(C(O)=O)C1=CC=CC=C1 FPPNZSSZRUTDAP-UWFZAAFLSA-N 0.000 description 1
- 229960003669 carbenicillin Drugs 0.000 description 1
- 235000020226 cashew nut Nutrition 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 229960004261 cefotaxime Drugs 0.000 description 1
- AZZMGZXNTDTSME-JUZDKLSSSA-M cefotaxime sodium Chemical compound [Na+].N([C@@H]1C(N2C(=C(COC(C)=O)CS[C@@H]21)C([O-])=O)=O)C(=O)\C(=N/OC)C1=CSC(N)=N1 AZZMGZXNTDTSME-JUZDKLSSSA-M 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 210000002421 cell wall Anatomy 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 229940124587 cephalosporin Drugs 0.000 description 1
- 150000001780 cephalosporins Chemical class 0.000 description 1
- 235000013339 cereals Nutrition 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 229930002868 chlorophyll a Natural products 0.000 description 1
- ATNHDLDRLWWWCB-AENOIHSZSA-M chlorophyll a Chemical compound C1([C@@H](C(=O)OC)C(=O)C2=C3C)=C2N2C3=CC(C(CC)=C3C)=[N+]4C3=CC3=C(C=C)C(C)=C5N3[Mg-2]42[N+]2=C1[C@@H](CCC(=O)OC\C=C(/C)CCC[C@H](C)CCC[C@H](C)CCCC(C)C)[C@H](C)C2=C5 ATNHDLDRLWWWCB-AENOIHSZSA-M 0.000 description 1
- 229930002869 chlorophyll b Natural products 0.000 description 1
- NSMUHPMZFPKNMZ-VBYMZDBQSA-M chlorophyll b Chemical compound C1([C@@H](C(=O)OC)C(=O)C2=C3C)=C2N2C3=CC(C(CC)=C3C=O)=[N+]4C3=CC3=C(C=C)C(C)=C5N3[Mg-2]42[N+]2=C1[C@@H](CCC(=O)OC\C=C(/C)CCC[C@H](C)CCC[C@H](C)CCCC(C)C)[C@H](C)C2=C5 NSMUHPMZFPKNMZ-VBYMZDBQSA-M 0.000 description 1
- 230000002759 chromosomal effect Effects 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000004590 computer program Methods 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 244000038559 crop plants Species 0.000 description 1
- 239000000287 crude extract Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 238000013480 data collection Methods 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000003412 degenerative effect Effects 0.000 description 1
- FCRACOPGPMPSHN-UHFFFAOYSA-N desoxyabscisic acid Natural products OC(=O)C=C(C)C=CC1C(C)=CC(=O)CC1(C)C FCRACOPGPMPSHN-UHFFFAOYSA-N 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- DENRZWYUOJLTMF-UHFFFAOYSA-N diethyl sulfate Chemical compound CCOS(=O)(=O)OCC DENRZWYUOJLTMF-UHFFFAOYSA-N 0.000 description 1
- 229940008406 diethyl sulfate Drugs 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000006471 dimerization reaction Methods 0.000 description 1
- MHUWZNTUIIFHAS-CLFAGFIQSA-N dioleoyl phosphatidic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(COP(O)(O)=O)OC(=O)CCCCCCC\C=C/CCCCCCCC MHUWZNTUIIFHAS-CLFAGFIQSA-N 0.000 description 1
- 238000002224 dissection Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- NAGJZTKCGNOGPW-UHFFFAOYSA-N dithiophosphoric acid Chemical class OP(O)(S)=S NAGJZTKCGNOGPW-UHFFFAOYSA-N 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000003337 fertilizer Substances 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 230000028245 fruit abscission Effects 0.000 description 1
- 239000003517 fume Substances 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 230000006251 gamma-carboxylation Effects 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 230000004545 gene duplication Effects 0.000 description 1
- 238000012252 genetic analysis Methods 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- 238000011331 genomic analysis Methods 0.000 description 1
- 229960002518 gentamicin Drugs 0.000 description 1
- 125000000291 glutamic acid group Chemical group N[C@@H](CCC(O)=O)C(=O)* 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 239000005090 green fluorescent protein Substances 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 230000033444 hydroxylation Effects 0.000 description 1
- 238000005805 hydroxylation reaction Methods 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000000415 inactivating effect Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000005865 ionizing radiation Effects 0.000 description 1
- 150000002576 ketones Chemical group 0.000 description 1
- 229960004502 levodopa Drugs 0.000 description 1
- 238000007834 ligase chain reaction Methods 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 230000013011 mating Effects 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 101150037888 mdv1 gene Proteins 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 1
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 239000011785 micronutrient Substances 0.000 description 1
- 235000013369 micronutrients Nutrition 0.000 description 1
- 239000003471 mutagenic agent Substances 0.000 description 1
- 231100000707 mutagenic chemical Toxicity 0.000 description 1
- 210000004897 n-terminal region Anatomy 0.000 description 1
- 101150091879 neo gene Proteins 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- NVGOPFQZYCNLDU-UHFFFAOYSA-N norflurazon Chemical compound O=C1C(Cl)=C(NC)C=NN1C1=CC=CC(C(F)(F)F)=C1 NVGOPFQZYCNLDU-UHFFFAOYSA-N 0.000 description 1
- YCIMNLLNPGFGHC-UHFFFAOYSA-N o-dihydroxy-benzene Natural products OC1=CC=CC=C1O YCIMNLLNPGFGHC-UHFFFAOYSA-N 0.000 description 1
- 238000006384 oligomerization reaction Methods 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 150000004713 phosphodiesters Chemical group 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 108010001545 phytoene dehydrogenase Proteins 0.000 description 1
- 229930195732 phytohormone Natural products 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 238000003976 plant breeding Methods 0.000 description 1
- 230000008635 plant growth Effects 0.000 description 1
- 229920000729 poly(L-lysine) polymer Polymers 0.000 description 1
- 230000001124 posttranscriptional effect Effects 0.000 description 1
- 230000001323 posttranslational effect Effects 0.000 description 1
- 230000009145 protein modification Effects 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 239000012521 purified sample Substances 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000004064 recycling Methods 0.000 description 1
- 238000003329 reductase reaction Methods 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 238000002271 resection Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 230000008117 seed development Effects 0.000 description 1
- 230000007226 seed germination Effects 0.000 description 1
- 238000005204 segregation Methods 0.000 description 1
- 239000006152 selective media Substances 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 230000009131 signaling function Effects 0.000 description 1
- 238000002791 soaking Methods 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 238000009987 spinning Methods 0.000 description 1
- 150000003432 sterols Chemical class 0.000 description 1
- 235000003702 sterols Nutrition 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 239000002438 stress hormone Substances 0.000 description 1
- YROXIXLRRCOBKF-UHFFFAOYSA-N sulfonylurea Chemical compound OC(=N)N=S(=O)=O YROXIXLRRCOBKF-UHFFFAOYSA-N 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 238000010257 thawing Methods 0.000 description 1
- 238000005382 thermal cycling Methods 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- 239000011573 trace mineral Substances 0.000 description 1
- 235000013619 trace mineral Nutrition 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- WCTAGTRAWPDFQO-UHFFFAOYSA-K trisodium;hydrogen carbonate;carbonate Chemical compound [Na+].[Na+].[Na+].OC([O-])=O.[O-]C([O-])=O WCTAGTRAWPDFQO-UHFFFAOYSA-K 0.000 description 1
- 230000007306 turnover Effects 0.000 description 1
- 101150101900 uidA gene Proteins 0.000 description 1
- 241001515965 unidentified phage Species 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 230000017260 vegetative to reproductive phase transition of meristem Effects 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 235000020234 walnut Nutrition 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 101150074257 xylE gene Proteins 0.000 description 1
- 238000004383 yellowing Methods 0.000 description 1
- 239000005019 zein Substances 0.000 description 1
- 229940093612 zein Drugs 0.000 description 1
- 239000011592 zinc chloride Substances 0.000 description 1
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/415—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from plants
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/82—Vectors or expression systems specially adapted for eukaryotic hosts for plant cells, e.g. plant artificial chromosomes (PACs)
- C12N15/8241—Phenotypically and genetically modified plants via recombinant DNA technology
- C12N15/8261—Phenotypically and genetically modified plants via recombinant DNA technology with agronomic (input) traits, e.g. crop yield
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A40/00—Adaptation technologies in agriculture, forestry, livestock or agroalimentary production
- Y02A40/10—Adaptation technologies in agriculture, forestry, livestock or agroalimentary production in agriculture
- Y02A40/146—Genetically Modified [GMO] plants, e.g. transgenic plants
Definitions
- This invention relates to plant molecular biology.
- this invention relates to transgenic plants having increased or enhanced seed and organ sizes.
- a plant goes through a development cycle, which includes seed germination, maturation of plant, reproduction, and finally senescence that leads to death of a plant.
- Several biological processes are common to different stages of plant development. Desired effects such as growth of tissue organ are achieved in nature by fine-tuning of the metabolism of the organism.
- the final phase of growth is senescence which is a highly regulated, genetically controlled and active process (Thomas H., and Stoddart J. L., Ann. Rev. Plant Physiol (31) 83- 111, 1980).
- Senescence is mostly studied in plant leaves and is regarded as a series of events concerned with cellular disassembly and the mobilization of released material to other plant parts such as seeds, storage organs or developing leaves and flowers (Nooden L.D. In Senescence and Aging in Plants, Academic press, 391-439, 1988).
- Leaf senescence can be initiated by seed development in certain species of plants. This was demonstrated in soybean by surgically removing flowers or physically restricting pod growth to observe the delay in leaf senescence (Nooden L.D. In Senescence and Aging in Plants, Academic press, 330-368,1988; Miceli F, Crafts-Brandner S.J., Egli D.B. Crop Sci. (35), 1080-1085, 1995).
- partitioning of resources between vegetative and reproductive development involves a complex interplay of generative and degenerative processes, requiring differential expression of genes.
- SAGs Synchronization Associated Genes
- the SAG 13 gene was first described by Lohman et al. in 1994 (Lohman K.M., Gan S.,
- the present invention relates to transgenic plants with increased organ size when compared to a non transformed plant of the same species.
- the present invention provides a transgenic plant with increased seed size when compared to a non transformed plant of the same species.
- One embodiment of the invention provides a method of increasing seed and organ size of a plant by transforming the plant with a DNA construct comprising a promoter that functions in plants, operably linked to a DNA molecule that encodes a protein, where in said DNA molecule is selected from the from the group consisting of SEQ ID NO: 1, SEQ ID NO: 3, SEQ ID NO: 5, SEQ ID NO: 7, SEQ ID NO: 9, SEQ ID NO: 11, SEQ ID NO: 13, SEQ ID NO: 15, SEQ ID NO: 17, SEQ ID NO: 19, SEQ ID NO: 21, SEQ ID NO: 23, SEQ ID NO: 25, SEQ ID NO: 27, SEQ ID NO: 29, SEQ ID NO: 31, SEQ ID NO: 33, SEQ ID NO: 35, SEQ ID NO: 37, SEQ ID NO: 39, SEQ ID NO: 41, SEQ ID NO: 43, SEQ ID NO: 45, SEQ ID NO: 47, SEQ ID NO: 49, SEQ ID NO: 51, SEQ ID NO: 53, SEQ ID NO: 66, S
- the method also includes the DNA construct operably linked to a 3' termination region and selecting a desired plant from a population of transformed plants containing said DNA construct; wherein said desired plant exhibits increased seed and organ size compared to a plant of a same plant species not transformed to contain said DNA construct.
- the method may also encompass various promoters including a caulimovirus promoter, or a heterologous plant constitutive promoter, or a tissue specific or an organ enhanced promoter.
- the present invention provides a method for increasing seed and other plant organ sizes by transforming a plant with a DNA construct comprising a promoter that functions in plants, operably linked to a DNA molecule that encodes a protein, wherein said protein comprises at least an N-terminal 50% portion of a polypeptide selected from the group consisting of : SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 6, SEQ ID NO: 8, SEQ ID NO: 10, SEQ ID NO: 12, SEQ ID NO: 14, SEQ ID NO: 16, SEQ ID NO: 18, SEQ ID NO: 20, SEQ ID NO: 22, SEQ ID NO: 24, SEQ ID NO: 26, SEQ ID NO: 28, SEQ ID NO: 30, SEQ ID NO: 32, SEQ ID NO: 34, SEQ ID NO: 36, SEQ ID NO: 38, SEQ ID NO: 40, SEQ ID NO: 42, SEQ ID NO: 44, SEQ ID NO: 46, SEQ ID NO: 48, SEQ ID NO: 50, SEQ ID NO: 52, SEQ
- the present invention provides an isolated DNA construct comprising a promoter capable of functioning in a plant cell, operably linked to a structural nucleic acid sequence encoding SEQ ID NO: 2 or SEQ ID NO: 4, and a 3' non translated nucleic acid sequence capable of causing transcriptional termination and the addition of polyadenylated nucleotides to the 3 'end of the transcribed mRNA sequence.
- This nucleic acid sequence may optionally include an intron, a 5' untranslated leader sequence or another nucleic acid sequence designed to enhance transcription and/or translation.
- Another aspect of the present invention provides a method for improving the seed and organ size in a plant comprising the steps of:
- SEQ ID NO: 22 SEQ ID NO: 24, SEQ ID NO: 26, SEQ ID NO: 28, SEQ ID NO: 30, SEQ ID NO: 32, SEQ ID NO: 34, SEQ ID NO: 36, SEQ ID NO: 38, SEQ ID NO: 40, SEQ ID NO: 42, SEQ ID NO: 44, SEQ ID NO: 46, SEQ ID NO: 48, SEQ ID NO: 50, SEQ ID NO: 52, SEQ ID NO: 54, SEQ ID NO: 67, SEQ ID NO: 69, and SEQ ID NO: 71 operably linked to
- step (b) obtaining transformed plant cells containing the nucleic acid of step (a);
- transgenic plant with increased seed and organ size when compared to a non transformed plant of a same plant species, said transgenic plant comprising a DNA construct, wherein said DNA construct comprises a promoter that functions in plants, operably linked to a DNA molecule that encodes a protein, wherein said DNA molecule is selected from the group consisting of: SEQ ID NO: 1, SEQ ID NO: 3, SEQ ID NO: 5, SEQ ID NO: 7, SEQ ID NO: 9, SEQ ID NO: 11, SEQ ID NO: 13, SEQ ID NO: 15, SEQ ID NO: 17, SEQ ID NO: 19, SEQ ID NO: 21, SEQ ID NO: 23, SEQ ID NO: 25, SEQ ID NO: 27, SEQ ID NO: 29, SEQ ID NO: 31, SEQ ID NO: 33, SEQ ID NO: 35, SEQ ID NO: 37, SEQ ID NO: 39, SEQ ID NO: 41, SEQ ID NO: 43, SEQ ID NO: 45, SEQ ID NO: 47, SEQ ID NO: 49, SEQ ID NO: 51,
- transgenic plant with increased seed and organ size when compared to a non transformed plant of a same plant species, the transgenic plant comprising a DNA construct, wherein said DNA construct encodes a protein that comprises at least an N-terminal 50% portion selected from the group consisting of: SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 6, SEQ ID NO: 8, SEQ ID NO: 10, SEQ ID NO: 12, SEQ ID NO: 14, SEQ ID NO: 16, SEQ ID NO: 18, SEQ ID NO: 20, SEQ ID NO: 22, SEQ ID NO: 24, SEQ ID NO: 26, SEQ ID NO: 28, SEQ ID NO: 30, SEQ ID NO: 32, SEQ ID NO: 34, SEQ ID NO: 36, SEQ ID NO: 38, SEQ ID NO: 40, SEQ ID NO: 42, SEQ ID NO: 44, SEQ ID NO: 46, SEQ ID NO: 48, SEQ ID NO: 50, SEQ ID NO: 52, SEQ ID NO: 54, SEQ ID NO: 67, S
- nucleotide sequence encodes a polypeptide having an amino acid sequence that has at least 50% sequence identity, preferably at least 60%, more preferably at least 70% sequence identity, even more preferably at least 80% or 90% sequence identity, and most preferably at least 95% to 98% sequence identity to a polypeptide selected from group consisting of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 6, SEQ ID NO: 8, SEQ ID NO: 10, SEQ ID NO: 12, SEQ ID NO: 14, SEQ ID NO: 16, SEQ ID NO: 18, SEQ ID NO: 20, SEQ ID NO: 22, SEQ ID NO: 24, SEQ ID NO: 26, SEQ ID NO: 28, SEQ ID NO: 30, SEQ ID NO: 32, SEQ ID NO: 34, SEQ ID NO: 36, SEQ ID NO: 38, SEQ ID NO: 40, SEQ ID NO:
- Figure 1 shows a plasmid map for plant transformation vector pMON 23435 with its elements.
- Figure 2 shows a plasmid map for plant transformation vector pMON 57521 with its elements.
- Figure 3 shows a plasmid map for plant transformation vector pMON 54955 with its elements.
- Figure 4 shows a plasmid map for plant transformation vector pMON 73955 with its elements.
- Figure 5 shows a plasmid map for plant transformation vector pMON 73963 with its elements.
- Figure 6 shows a plasmid map for bacterial transformation vector pMON 63132 with its elements.
- Figure 7 shows a plasmid map for bacterial transformation vector pMON 63133 with its elements.
- Figure 8 shows a plasmid map for bacterial transformation vector pMON 6134 with its elements.
- Figure 9 shows a plasmid map for bacterial transformation vector pMON 6135 with its elements.
- Figure 10 shows primers used for amplification of gene and for sequencing of amplicons.
- Figure 11 shows putative products from steroid reductase activity.
- Figure 12 shows a potential steroid reductase reaction.
- the present invention provides a method of increasing seed and organ size of a plant by transforming the plant with a DNA construct comprising a promoter that functions in plants, operably linked to a DNA molecule that encodes a protein, where in said DNA molecule is selected from the from the group consisting of SEQ ID NO: 1, SEQ ID NO: 3, SEQ ID NO: 5, SEQ ID NO: 7, SEQ ID NO: 9, SEQ ID NO: 11, SEQ ID NO: 13, SEQ ID NO: 15, SEQ ID NO: 17, SEQ ID NO: 19, SEQ ID NO: 21, SEQ ID NO: 23, SEQ ID NO: 25, SEQ ID NO: 27, SEQ ID NO: 29, SEQ ID NO: 31, SEQ ID NO: 33, SEQ ID NO: 35, SEQ ID NO: 37, SEQ ID NO: 39, SEQ ID NO: 41, SEQ ID NO: 43, SEQ ID NO: 45, SEQ ID NO: 47, SEQ ID NO: 49, SEQ ID NO: 51, SEQ ID NO: 53, SEQ ID NO: 66, SEQ ID
- the present invention provides a method for increasing seed and other plant organ sizes by transforming a plant with a DNA construct comprising a promoter that functions in plants, operably linked to a DNA molecule that encodes a protein, wherein said protein comprises at least an N-terminal 50% portion of a polypeptide selected from the group consisting of : SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 6, SEQ ID NO: 8, SEQ ID NO: 10, SEQ ID NO: 12, SEQ ID NO: 14, SEQ ID NO: 16, SEQ ID NO: 18, SEQ ID NO: 20, SEQ ID NO: 22, SEQ ID NO: 24, SEQ ID NO: 26, SEQ ID NO: 28, SEQ ID NO: 30, SEQ ID NO: 32, SEQ ID NO: 34, SEQ ID NO: 36, SEQ ID NO: 38, SEQ ID NO: 40, SEQ ID NO: 42, SEQ ID NO: 44, SEQ ID NO: 46, SEQ ID NO: 48, SEQ ID NO: 50, SEQ ID NO: 52, SEQ
- nucleic acid molecule as used herein means a deoxyribonucleic acid (DNA) molecule or ribonucleic acid (RNA) molecule. Both DNA and RNA molecules are constructed from nucleotides linked end to end, wherein each of the nucleotides contains a phosphate group, a sugar moiety, and either a purine or a pyrimidine base. Nucleic acid molecules can be a single or double-stranded polymer of nucleotides read from the 5' to the 3' end. Nucleic acid molecules may also optionally contain synthetic, non-natural or altered nucleotide bases that permit correct read through by a polymerase and do not alter expression of a polypeptide encoded by that nucleic acid molecule.
- an isolated nucleic acid molecule means a nucleic acid molecule that is no longer accompanied by some of materials with which it is associated in its natural state or to a nucleic acid molecule the structure of which is not identical to that of any of naturally occurring nucleic acid molecule.
- an isolated nucleic acid molecule examples include: (1) DNAs which have the sequence of part of a naturally occurring genomic DNA molecule but are not flanked by two coding sequences that flank that part of the molecule in the genome of the organism in which it naturally occurs; (2) a nucleic acid molecule incorporated into a vector or into the genomic DNA of a prokaryote or eukaryote in a manner such that the resulting molecule is not identical to any naturally occurring vector or genomic DNA; (3) a separate molecule such as a cDNA, a genomic fragment, a fragment produced by polymerase chain reaction (PCR), or a restriction fragment; (4) recombinant DNAs; and (5) synthetic DNAs.
- An isolated nucleic acid molecule may also be comprised of one or more segments of cDNA, genomic DNA or synthetic DNA.
- the isolated nucleic acid molecules of the present invention also include known types of modifications, for example, labels which are known in the art, methylation, "caps", substitution of one or more of the naturally occurring nucleotides with an analog.
- internucleotide modifications for example, those with uncharged linkages (methyl phosphonates, phosphotriesters, phosphoamidates, carbamates, etc.) and with charged linkages (phosphorothioates, phosphorodithioates, etc.), those containing pendant moieties, such as, proteins (including nucleases, toxins, antibodies, signal peptides, poly-L-lysine, etc.), those with intercalators (acridine, psoralen, etc.), those containing chelators (metals, radioactive metals, boron, oxidative metals, etc.), those containing alkylators, and those with modified linkages.
- uncharged linkages methyl phosphonates, phosphotriesters, phosphoamidates, carbamates, etc.
- charged linkages phosphorothioates, phosphorodithioates, etc.
- pendant moieties such as, proteins (including nucleases, toxins, antibodies, signal peptides, poly-
- nucleotide sequence means the order contiguous of nucleic acid molecule of both the sense and antisense strands or as a duplex strand. It includes, but is not limited to, self-replicating plasmids, synthetic polynucleotides, chromosomal sequences, and infectious polymers of DNA or RNA.
- a nucleotide sequence is said to be the "complement” of another nucleotide sequence if they exhibit complete complementarity.
- molecules are said to exhibit "complete complementarity" when every nucleotide of one of the sequences is complementary to a nucleotide of the other.
- a coding sequence and "a structural nucleotide sequence” mean a nucleotide sequence, which is translated into a polypeptide, usually via mRNA, when placed under the control of appropriate regulatory sequences. The boundaries of the coding sequence are determined by a translation start codon at the 5 '-terminus and a translation stop codon at the 3 '-terminus.
- a coding sequence can include, but is not limited to, genomic DNA, cDNA, and recombinant nucleotide sequences.
- polypeptides of the invention like other polypeptides, have different domains which perfo ⁇ n different functions. Thus, the coding sequences need not be full length, so long as the desired functional domain of the polypeptide is expressed.
- the distinguishing features of polypeptides of the present invention are discussed in detail in Examples.
- the term "recombinant DNAs" as used herein means DNAs that contains a genetically engineered modification through manipulation via mutagenesis, restriction enzymes, and the like.
- synthetic DNAs as used herein means DNAs assembled from oligonucleotide building blocks that are chemically synthesized using procedures known to those skilled in the art. These building blocks are ligated and annealed to form DNA segments, which are then enzymatically assembled to construct the entire DNA. "Chemically synthesized”, as related to a sequence of DNA, means that the component nucleotides were assembled in vitro. Manual chemical synthesis of DNA may be accomplished using well-established procedures, or automated chemical synthesis can be performed using one of a number of commercially available machines.
- polypeptide and protein mean a polymer composed of amino acids connected by peptide bonds.
- An amino acid unit in a polypeptide (or protein) is called a residue.
- polypeptide and protein also applies to any amino acid polymers in which one or more amino acid residue is an artificial chemical analogue- of a corresponding naturally occurring amino acid, as well as to any naturally occurring amino acid polymers.
- the essential nature of such analogues of naturally occurring amino acids is that, when incorporated into a polypeptide, that polypeptide is specifically reactive to antibodies elicited to the same polypeptide but consisting entirely of naturally occurring amino acids.
- proteins or polypeptides may undergo modification, including but not limited to, disulfide bond formation, gamma-carboxylation of glutamic acid residues, glycosylation, lipid attachment, phosphorylation, oligomerization, hydroxylation and ADP-ribosylation.
- modification including but not limited to, disulfide bond formation, gamma-carboxylation of glutamic acid residues, glycosylation, lipid attachment, phosphorylation, oligomerization, hydroxylation and ADP-ribosylation.
- Exemplary modifications are described in most basic texts, such as, for example, Proteins - Structure and Molecular Properties, 2nd ed., T. E. Creighton, W. H. Freeman and Company, New York (1993), herein inco ⁇ orated by reference in its entirety. Many detailed reviews are available on this subject, such as, for example, those provided by Wold, F., Post-translational Protein Modifications. Perspectives and Prospects, pp.
- polypeptides blockages of the amino or carboxyl group in a polypeptide, or both, by a covalent modification, is common in naturally occurring and synthetic polypeptides and such modifications may be present in polypeptides of the present invention, as well.
- a methionine residue at the NH 2 terminus may be deleted.
- this invention contemplates the use of both the methionine containing and the methionine-less amino terminal variants of the polypeptide of the invention.
- amino acid and “amino acids” refer to all naturally occurring amino acids and, unless otherwise limited, known analogs of natural amino acids that can function in a similar manner as naturally occurring amino acids. This definition is meant to include ⁇ norleucine, ornithine, homocysteine, and homoserine.
- amino acid sequence means the sequence of amino acids in a polypeptide (or protein) that is written starting with the amino-terminal (N-terminal) residue and ending with the carboxyl-terminal (C-terminal) residue.
- an amino acid subsequence means a portion of the amino acid sequence of a polypeptide.
- An amino acid subsequence generally has a length of 3 to 50 amino acid residues.
- substantially purified polypeptide and “substantially purified protein”, as used herein, means a polypeptide or protein that is separated substantially from all other molecules normally associated with it in its native state and is the predominant species present in a preparation.
- a substantially purified molecule may be greater than 60% free, preferably 75% free, more preferably 90% free, and most preferably 95% free from the other molecules (exclusive of solvent) present in the natural mixture.
- Percentage of sequence identity is determined by comparing two optimally aligned sequences over a comparison window, wherein the portion of the polynucleotide or amino acid sequence in the comparison window may comprise additions or deletions (i.e., gaps) as compared to the reference sequence (which does not comprise additions or deletions) for optimal alignment of the two sequences.
- the percentage is calculated by determining the number of positions at which the identical nucleic acid base or amino acid residue occurs in both sequences to yield the number of matched positions, dividing the number of matched positions by the total number of positions in the window of comparison and multiplying the result by 100 to yield the percentage of sequence identity.
- identity refers to amino acid or nucleic acid sequences that when compared using the local homology algorithm of Smith and Waterman (T. F. Smith and M. S. Waterman, J. Mol. Biol. (147) 195-197 (1981). in the SSEARCH3 3.0t75 (W.R. Pearson, Genomics (11)635- 650 (1991) or BLAST 2.2.1 (Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman , Nucleic Acids Res. (25)3389- 3402 (1997) programs which are exactly alike.
- similarity refers to two amino acids, which are similar as defined by similarity matrix BLOSSUM62 (S. Henikoff and J. G. Henikoff, , Proc. Natl. Acad. Sci. U.S.A. (89) 10915-10919 (1992) which is used in BLAST 2.2.1 (Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman , Nucleic Acids Res. (25)3389-3402 (1997).
- the BLAST uses alias "positive” for similarity.
- percent identity for a pair of protein sequences refers to the number of identical amino acid residues in a two-sequence alignment reported by BLAST, divided by the total number of amino acid residues in the same alignment, expressed in percentage.
- percent similarity for a pair of protein sequences refers to the number of similar (“Positive” in BLAST output) amino acid residues in a two-sequence alignment reported by BLAST, divided by the total number of amino acid residues in the same alignment, expressed in percentage.
- Polypeptides which are "substantially similar” share sequences as noted above except that residue positions which are not identical may differ by conservative amino acid changes.
- Conservative amino acid substitutions refer to the interchangeability of residues having similar side chains.
- Constant amino acid substitutions mean substitutions of one or more amino acids in a native amino acid sequence with another amino acid(s) having similar side chains, resulting in a silent change. conserveed substitutes for an amino acid within a native amino acid sequence can be selected from other members of the group to which the naturally occurring amino acid belongs.
- a group of amino acids having aliphatic side chains is glycine, alanine, valine, leucine, and isoleucine; a group of amino acids having aliphatic-hydroxyl side chains is serine and threonine; a group of amino acids having amide-containing side chains is asparagine and glutamine; a group of amino acids having aromatic side chains is phenylalanine, tyrosine, and tryptophan; a group of amino acids having basic side chains is lysine, arginine, and histidine; and a group of amino acids having sulfur-containing side chains is cysteine and methionine.
- Preferred conservative amino acids substitution groups are: valine-leucine, valine- isoleucine, phenylalanine-tyrosine, lysine-arginine, alanine-valine, aspartic acid-glutamic acid, and asparagine-glutamine.
- nucleotide sequences can be appropriately adjusted to determine corresponding sequence identity of two nucleotide sequences encoding the polypeptides of the present invention by taking into account codon degeneracy, conservative amino acid substitutions, reading frame positioning and the like. Substantial identity of nucleotide sequences for these purposes normally means sequence identity of at least 35%).
- codon degeneracy means divergence in the genetic code permitting variation of the nucleotide sequence without affecting the amino acid sequence of an encoded polypeptide.
- codon-bias exhibited by a specific host cell in usage of nucleotide codons to specify a given amino acid.
- the present invention provides an isolated nucleic acid molecule in a DNA construct comprising a promoter operably linked to nucleotide sequence or complement thereof, wherein the nucleotide sequence hybridizes under stringent conditions to the complement of a second nucleotide sequence encoding a polypeptide having an amino acid sequence selected from the group consisting of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 6, SEQ ID NO: 8, SEQ ID NO: 10, SEQ ID NO: 12, SEQ ID NO: 14, SEQ ID NO: 16, SEQ ID NO: 18, SEQ ID NO: 20, SEQ ID NO: 22, SEQ ID NO: 24, SEQ ID NO: 26, SEQ ID NO: 28, SEQ ID NO: 30, SEQ ID NO: 32, SEQ ID NO: 34, SEQ ID NO: 36, SEQ ID NO:
- the present invention also provides a method for obtaining an isolated nucleic acid molecule of the present invention, the method comprising the steps of: (a) probing a cDNA or genomic library with a hybridization probe comprising a nucleotide sequence encoding all or a portion of the amino acid sequence of a polypeptide, wherein the amino acid sequence of the polypeptide is selected from the group consisting of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 34, SEQ ID NO: 36, SEQ ID NO: 38, SEQ ID NO: 40, SEQ ID NO: 42, SEQ ID NO: 44, SEQ ID NO: 46, SEQ ID NO: 48, SEQ ID NO: 52, SEQ ID NO: 54, SEQ ID NO: 67, SEQ ID NO: 69, and SEQ ID NO: 71 (b) identifying a DNA clone that hybridizes under stringent conditions to hybridization probe; (c) isolating the DNA clone identified in step (b); and (d)
- Hybridization conditions are sequence dependent and will be different in different circumstances.
- stringent conditions are selected to be about 5°C lower than the thermal melting point (Tm) for the specific sequence at a defined ionic strength and pH.
- the "thermal melting point” is the temperature (under defined ionic strength and pH) at which 50% of a target molecule hybridizes to a completely complementary molecule.
- Appropriate stringent conditions which promote DNA hybridization for example, 6.0 X sodium chloride/sodium citrate (SSC) at about 45°C, followed by a wash of 2.0 X SSC at 50°C, are known to those skilled in the art or can be found in Current Protocols in Molecular Biology, John Wiley & Sons, N.Y.
- the salt concentration in the wash step can be selected from a low stringent condition of about 2.0 X SSC at 50°C to a high stringency of about 0.2 X SSC at 50°C.
- the temperature in the wash step can be increased from low stringent conditions at room temperature, about 22°C, to high stringent conditions at about 65°C. Both temperature and salt concentration may be varied, or either the temperature or the salt concentration may be held constant while the other variable is changed.
- stringent conditions include at least one wash in 2.0 X SSC at a temperature of at least about 50°C for 20 minutes, or equivalent conditions.
- nucleic acid molecules of the present invention may be combined with other non- native, or “heterologous” sequences in a variety of ways.
- heterologous sequences it is meant any sequence that is not naturally found joined to the nucleotide sequence encoding polypeptide of the present invention, including, for example, combinations of nucleotide sequences from the same plant which are not naturally found joined together, or the two sequences originate from two different species.
- homologous refers to two or more genes that are derived from a single gene in a common ancestor.
- homoology is attributed to decent from a common ancestor.
- orthologous refers to different homologous sequences in different species that arose from a common ancestral gene during speciation; may or may not be responsible for a similar function.
- paralogous refers to homologous sequences within a single species that arose by gene duplication.
- domain refers to a discrete portion of a protein assumed to fold independently of the rest of the protein and possessing its own function. Domain is also used to refer a discrete portion of nucleotide when translated provides a portion of protein which is assumed to fold independently of the rest of the translated nucleotide sequence and possessing its own function.
- Motif refers to a short conserved region in a protein or nucleotide sequence. Motifs are frequently highly conserved parts of domains.
- the term "genome” as it applies to plant cells encompasses not only chromosomal DNA found within the nucleus, but organelle DNA found within subcellular components of the cell. DNAs of the present invention introduced into plant cells can therefore be either chromosomally integrated or organelle-localized.
- operably linked as used in reference to a regulatory sequence and a structural nucleotide sequence, means that the regulatory sequence causes regulated expression of the operably linked structural nucleotide sequence.
- “Expression” means the transcription and stable accumulation of sense or antisense RNA derived from the nucleic acid molecule of the present invention. Expression may also refer to translation of mRNA into a polypeptide.
- RNA transcript means RNA transcript that includes the mRNA and so can be translated into polypeptide or protein by the cell.
- Antisense RNA means a RNA transcript that is complementary to all or part of a target primary transcript or mRNA and that blocks the expression of a target gene (U.S. Pat. No. 5,107,065, incorporated herein by reference). The complementarity of an antisense RNA may be with any part of the specific gene transcript, i.e., at the 5' non-coding sequence, 3' non- translated sequence, introns, or the coding sequence.
- RNA transcript means the product resulting from RNA polymerase-catalyzed transcription of a DNA sequence. When the RNA transcript is a perfect complementary copy of the DNA sequence, it is referred to as the primary transcript or it may be a RNA sequence derived from post-transcriptional processing of the primary transcript and is referred to as the mature RNA.
- overexpression means the expression of a polypeptide encoded by an exogenous nucleic acid molecule introduced into a host cell, wherein said polypeptide is either not normally present in the host cell, or wherein said polypeptide is present in said host cell at a higher level than that normally expressed from the endogenous gene encoding said polypeptide.
- ectopic expression it is meant that expression of a nucleic acid molecule encoding a polypeptide in a cell type other than a cell type in which the nucleic acid molecule is normally expressed, at a time other than a time at which the nucleic acid molecule is normally expressed or at a expression level other than the level at which the nucleic acid molecule normally is expressed.
- Antisense inhibition means the production of antisense RNA transcripts capable of suppressing the expression of the target polypeptide.
- Co-suppression means the production of sense RNA transcripts capable of suppressing the expression of identical or substantially similar foreign or endogenous genes (U.S. Patent No. 5,231,020, incorporated herein by reference).
- a gene means the segment of DNA that is involved in producing a polypeptide. Such segment of DNA includes regulatory sequences preceding (5' non-coding sequences) and following (3' non-coding sequences) the coding region as well as intervening sequences (introns) between individual coding segments (exons).
- a "Native gene” means a gene as found in nature with its own regulatory sequences.
- Chimeric gene means any gene that is not a native gene, comprising regulatory and coding sequences that are not found together in nature. Accordingly, a chimeric gene may comprise regulatory sequences and coding sequences that are derived from different sources, or regulatory sequences and coding sequences derived from the same source, but arranged in a manner different than that found in nature.
- Endogenous gene or "endogenous DNA molecule” means a native gene in its natural location in the genome of an organism.
- a “foreign gene” means a gene not normally found in the host organism, but that is introduced into the host organism by gene transfer.
- Foreign genes can comprise native genes inserted into a non-native organism, or chimeric genes.
- a “transgene” is a gene that has been introduced into the genome by a transformation procedure.
- Regulatory sequences mean nucleotide sequences located upstream (5' non-coding sequences), within, or downstream (3' non-translated sequences) of a structural nucleotide sequence, and which influence the transcription, RNA processing or stability, or translation of the associated structural nucleotide sequence. Regulatory sequences may include promoters, translation leader sequences, introns, and polyadenylation recognition sequences.
- promoter sequence means a nucleotide sequence that is capable of, when located in cis to a structural nucleotide sequence encoding a polypeptide, functioning in a way that directs expression of one or more mRNA molecules that encodes the polypeptide. Such promoter regions are typically found upstream of the trinucleotide ATG sequence at the start site of a polypeptide-coding region. Promoter sequences can also include sequences from which transcription of transfer RNA (tRNA) or ribosomal RNA (rRNA) sequences are initiated. Transcription involves the synthesis of a RNA chain representing one strand of a DNA duplex.
- tRNA transfer RNA
- rRNA ribosomal RNA
- RNA polymerase By “representing” it is meant that the RNA is identical in sequence with one strand of the DNA; it is complementary to the other DNA strand, which provides the template for its synthesis. Transcription takes place by the usual process of complementary base pairing, catalyzed and scrutinized by the enzyme RNA polymerase. The reaction can be divided into three stages described as initiation, elongation and termination. Initiation begins with the binding of RNA polymerase to the double stranded (DS or ds) DNA. The sequence of DNA required for the initiation reaction defines the promoter. The site at which the first nucleotide is incorporated is called the start-site or start-point of transcription. Elongation describes the phase during which the enzyme moves along the DNA and extends the growing RNA chain.
- Elongation involves the disruption of the DNA double stranded structure in which a transiently unwound region exists as a hybrid RNA-DNA duplex and a displaced single strand of DNA. Termination involves recognition of the point at which no further bases should be added to the chain. To terminate transcription, the formation of phosphodiester bonds must cease and the transcription complex must come apart. When the last base is added to the RNA chain, the RNA-DNA hybrid is disrupted, the DNA reforms into a duplex state, and the RNA polymerase enzyme and RNA molecule are both released from the DNA. The sequence of DNA required for the termination reaction is called the terminator.
- the promoter sequence consists of proximal and more distal upstream elements, the latter elements often referred to as enhancers.
- an “enhancer” is a DNA sequence that can stimulate promoter activity and may be an innate element of the promoter or a heterologous element inserted to enhance the level or tissue-specificity of a promoter. Promoters may be derived in their entirety from a native gene, or be composed of different elements derived from different promoters found in nature, or even comprise synthetic DNA segments. It is understood by those skilled in the art that different promoters may direct the expression of a gene in different tissues or cell types, or at different stages of development, or in response to different environmental conditions.
- Promoters which are known or are found to cause transcription of DNA in plant cells, can be used in the present invention. Such promoters may be obtained from a variety of sources such as plants and plant viruses. A number of promoters, including constitutive promoters, inducible promoters and tissue-specific promoters, that are active in plant cells have been described in the literature. In addition to promoters that are known to cause transcription of DNA in plant cells, other promoters may be identified for use in the current invention by screening a plant cDNA library for genes that are selectively or preferably expressed in the target tissues and then determine the promoter regions.
- constitutive promoter means a regulatory sequence which causes expression of a structural nucleotide sequence in most cells or tissues at most times. Constitutive promoters are active under most environmental conditions and states of development or cell differentiation. A variety of constitutive promoters are well known in the art.
- constitutive promoters that are active in plant cells include but are not limited to the nopaline synthase (NOS) promoters; DNA plant virus promoters including, but not limited to the caulimovirus promoters for example, cauliflower mosaic virus (CaMV) 19S and 35S, and figwort mosaic virus promoters; the bacilliform virus promoter for example sugar cane bacilliform virus, rice tungro bacilliform virus, among others; plant actin promoters, such as the Arabidopsis and rice actin gene promoter (see, e.g., Huang et al, Plant Mol. Biol. 33:125-139 (1997), US Patent No. 5,641,876), herein incorporated by reference in its entirety). These promoters when used in a DNA construct are heterologous to the linked gene sequence when they are derived from a different organism, plant species, or a different gene.
- NOS nopaline synthase
- DNA plant virus promoters including, but not limited to the caul
- inducible promoter means a regulatory sequence which causes conditional expression of a structural nucleotide sequence under the influence of changing environmental conditions or developmental conditions.
- inducible promoters include but are not limited to the senescence-induced promoter for the senescence-associated gene, SAG 12, (Gan and Amasino, Science 270: 1986-1988 (1995), herein incorporated by reference in its entirety); the light-inducible promoter from the small subunit of ribulose-l,5-bis-phosphate carboxylase (ssRUBISCO); the drought-inducible promoter of maize (Busk et al, Plant J.
- auxin-response elements El promoter fragment (AuxREs) in the soybean (Glycine max L.) (Liu et al, Plant Physiol. 775:397-407 (1997), herein incorporated by reference in its entirety); the auxin-responsive Arabidopsis GST6 promoter (also responsive to salicylic acid and hydrogen peroxide) (Chen et al, Plant J. 10: 955-966 (1996), herein inco ⁇ orated by reference in its entirety); the auxin- inducible parC promoter from tobacco (Sakai et al, Plant Cell Physiol.
- tissue-specific promoter or "organ enhanced promoter” means a regulatory sequence that causes transcriptions or enhanced transcriptions of DNA in specific cells or tissues at specific times during plant development, such as in vegetative tissues or reproductive tissues.
- tissue-specific promoters under developmental control include promoters that initiate transcription only (or primarily only) in certain tissues, such as vegetative tissues, e.g., roots, leaves or stems, or reproductive tissues, such as fruit, ovules, seeds, pollen, pistols, flowers, or any embryonic tissue.
- Reproductive tissue specific promoters may be, e.g., ovule- specific, embryo-specific, endosperm-specific, integument-specific, seed coat-specific, pollen- specific, petal-specific, sepal-specific, or some combination thereof.
- tissue-specific promoter may drive expression of operably linked sequences in tissues other than the target tissue.
- a tissue-specific promoter is one that drives expression preferentially in the target tissue, but may also lead to some expression in other tissues as well.
- tuber-specific promoters include but are not limited to the class I and II patatin promoters (Bevan et al, EMBO J. 8: 1899-1906 (1986); Koster-Topfer et al, Mol Gen Genet. 219: 390-396 (1989); Mignery et al, Gene. 62: 27-44 (1988); Jefferson et al, Plant Mol.
- leaf-specific promoters include but are not limited to the ribulose biphosphate carboxylase (RBCS or RuBISCO) promoters (see, e.g., Matsuoka et al, Plant J. 6:311-319 (1994), herein inco ⁇ orated by reference in its entirely); the light harvesting chlorophyll a/b binding protein gene promoter (see, e.g., Shhna et ⁇ /., R/ ⁇ ..tR/nw ⁇ /. 775:477-483 (1997); Cass ⁇ et al, Plant Physiol.
- RBCS or RuBISCO ribulose biphosphate carboxylase
- Root-specific promoter examples include but are not limited to the promoter for the acid chitinase gene (Samac et al, Plant Mol. Biol 25: 587-596 (1994), herein inco ⁇ orated by reference in its entirety); the root specific subdomains of the CaMV35S promoter that have been identified (Lam et al, Proc. Natl. Acad. Sci.
- Another class of useful vegetative tissue-specific promoters is meristematic (root tip and shoot apex) promoters.
- meristematic (root tip and shoot apex) promoters For example, the "SHOOTMERISTEMLESS” and “SCARECROW” promoters, which are active in the developing shoot or root apical meristems (Di Laurenzio et al, Cell 86:423- 433 (1996); Long, Nature 379:66-69 (1996); herein inco ⁇ orated by reference in their entireties), can be used.
- Another example of a useful promoter is that which controls the expression of 3-hydroxy-3- methylglutaryl coenzyme A reductase HMG2 gene, whose expression is restricted to meristematic and floral (secretory zone of the stigma, mature pollen grains, gynoecium vascular tissue, and fertilized ovules) tissues (see, e.g., Enjuto et al, Plant Cel 7:517-527 (1995), herein incorporated by reference in its entirety).
- Another example of a useful promoter is that which controls the expression of knl-related genes from maize and other species which show meristem-specific expression (see, e.g., Granger et al, Plant Mol Biol.
- KNAT1 Arabidopsis thaliana KNAT1 promoter.
- KNAT1 transcript is localized primarily to the shoot apical meristem; the expression of KNATI in the shoot meristem decreases during the floral transition and is restricted to the cortex of the inflorescence stem (see, e.g., Lincoln et al, Plant Cell
- Suitable seed-specific promoters can be derived from the following genes: MAC1 from maize (Sheridan et al, Genetics 742:1009-1020 (1996), herein inco ⁇ orated by reference in its entirety); Cat3 from maize (GenBank No. L05934, Abler et al, Plant Mol. Biol. 22:10131-1038
- GenBank No. U39944; Ray et al, Proc. Natl. Acad. Sci. USA 91:5161-5165 (1994), all of which are herein incorporated by reference in their entireties) can also be used.
- the egg and central cell specific MEA (FIS1) and FIS2 promoters are also useful reproductive tissue-specific promoters (Luo et al., Proc. Natl. Acad. Sci. USA, 97:10637-10642 (2000); Dahlle-Calzada, et al., Genes
- a maize pollen-specific promoter has been identified in maize (Guerrero et al., Mol. Gen.
- Promoters derived from genes encoding embryonic storage proteins which includes the gene encoding the 2S storage protein from Brassica napus (Dasgupta et al, Gene 133:301-302 (1993), herein incorporated by reference in its entirety); the 2S seed storage protein gene family from Arabidopsis; the gene encoding oleosin 20kD from Brassica napus (GenBank No.
- Promoters derived from zein encoding genes can be also used.
- the zeins are a group of storage proteins found in maize endosperm.
- promoters known to function include the promoters for the following genes: waxy, Brittle, Shrunken 2, Branching enzymes I and II, starch synthases, debranching enzymes, oleosins, glutelins, and sucrose synthases.
- a particularly preferred promoter for maize endosperm expression is the promoter for the glutelin gene from rice, more particularly the Osgt-1 promoter (Zheng et al, Mol. Cell Biol. 13: 5829-5842 (1993), herein inco ⁇ orated by reference in its entirety).
- promoters suitable for expression in wheat include those promoters for the ADPglucose pyrophosphorylase (ADPGPP) subunits, the granule bound and other starch synthases, the branching and debranching enzymes, the embryogenesis-abundant proteins, the gliadins, and the glutenins.
- promoters in rice include those promoters for the ADPGPP subunits, the granule bound and other starch synthases, the branching enzymes, the debranching enzymes, sucrose synthases, and the glutelins.
- a particularly preferred promoter is the promoter for rice glutelin, Osgt-1.
- promoters for barley include those for the ADPGPP subunits, the granule bound and other starch synthases, the branching enzymes, the debranching enzymes, sucrose synthases, the hordeins, the embryo globulins, and the aleurone specific proteins.
- a tomato promoter active during fruit ripening, senescence and abscission of leaves and, to a lesser extent, of flowers can be used (Blume et al., Plant J. 72:731-746 (1997), herein incorporated by reference in its entirety).
- Other exemplary promoters include the pistol specific promoter in the potato (Solarium tuberosum L.) SK2 gene, encoding a pistil-specific basic endochitinase (Ficker et al, Plant Mol Biol. 35:425-431 (1997), herein inco ⁇ orated by reference in its entirety); the Blec4 gene from pea (Pisum sativum cv.
- tissue specific E8 promoter from tomato is also useful for directing gene expression in fruits. It is recognized that additional promoters that may be utilized are described, for example, in U.S. Patent Nos. 5,378,619, 5,391,725, 5,428,147, 5,447,858, 5,608,144, 5,608,144, 5,614,399, 5,633,441, 5,633,435, and 4,633,436, all of which are herein inco ⁇ orated in their entirety.
- tissue specific enhancer may be used (Fromm et al., The Plant Cell 7:977-984 (1989), herein inco ⁇ orated by reference in its entirety). It is further recognized that since in most cases the exact boundaries of regulatory sequences have not been completely defined, DNA fragments of different lengths may have identical promoter activity.
- the "translation leader sequence” means a DNA sequence located between the promoter sequence of a gene and the coding sequence.
- the translation leader sequence is present in the fully processed mRNA upstream of the translation start sequence.
- the translation leader sequence may affect processing of the primary transcript to mRNA, mRNA stability or translation efficiency. Examples of translation leader sequences have been described (US Patent No. 5,659,122 and Turner and Foster, Molecular Biotechnology 3:225 (1995) herein inco ⁇ orated by reference in its entirety).
- the "3' non-translated sequences” means DNA sequences located downstream of a structural nucleotide sequence and include sequences encoding polyadenylation and other regulatory signals capable of affecting mRNA processing or gene expression.
- the polyadenylation signal functions in plants to cause the addition of polyadenylate nucleotides to the 3' end of the mRNA precursor.
- the polyadenylation sequence can be derived from the natural gene, from a variety of plant genes, or from T-DNA.
- An example of the polyadenylation sequence is the nopaline synthase 3' sequence (NOS 3'; Fraley et al, Proc. Natl. Acad. Sci. USA 80: 4803-4807 (1983), herein inco ⁇ orated by reference in its entirety).
- NOS 3' Fraley et al, Proc. Natl. Acad. Sci. USA 80: 4803-4807 (1983
- the use of different 3' non-translated sequences is exemplified by Ingelbrecht et al, Plant Cell 7:671-680 (1989), herein inco ⁇ orated by reference in its entirety.
- the isolated nucleic acid molecules of the present invention may also include introns.
- an intron sequence is inserted between the promoter sequence and the structural gene sequence or, optionally, may be inserted in the structural coding sequence to provide an interrupted coding sequence.
- An example of such an intron sequence is the HSP 70 intron described in WO 93/19189, herein inco ⁇ orated by reference in its entirety.
- the laboratory procedures in recombinant DNA technology used herein are those well known and commonly employed in the art. Standard techniques are used for cloning, DNA and RNA isolation, amplification and purification. Generally enzymatic reactions involving DNA ligase, DNA polymerase, restriction endonucleases and the like are performed according to the manufacturer's specifications. These techniques and various other techniques are generally performed according to Sambrook et al., Molecular Cloning - A Laboratory Manual, 2nd. ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, New York (1989).
- nucleic acid molecules encoding polypeptides of the present invention from soybean, corn, rice and other species are described in detail in Examples. All or a substantial portion of the nucleic acid molecules of the present invention may be used to isolate cDNAs and nucleic acid molecules encoding homologous polypeptides from the same or other plant species.
- a "substantial portion" of a nucleotide sequence comprises enough of the sequence to afford specific identification and/or isolation of a nucleic acid molecule comprising the sequence.
- Nucleotide sequences can be evaluated either manually by one skilled in the art, or by using computer-based sequence comparison and identification tools that employ algorithms such as BLAST (Basic Local Alignment Search Tool; Altschul et al. JMol Biol 275:403-410 (1993). In general, a sequence of thirty or more contiguous nucleotides is necessary in order to putatively identify a nucleotide sequence as homologous to a gene.
- gene-specific oligonucleotide probes comprising 30 or more contiguous nucleotides may be used in sequence-dependent methods of gene identification (e.g., Southern hybridization) and isolation (e.g., in situ hybridization of bacterial colonies or bacteriophage plaques).
- short oligonucleotides of 12 or more nucleotides may be used as amplification primers in PCR in order to obtain a particular nucleic acid molecule comprising the primers.
- sequence-dependent protocols include, but are not limited to, methods of nucleic acid molecule hybridization, and methods of DNA and RNA amplification as exemplified by various uses of nucleic acid molecule amplification technologies (e.g., polymerase chain reaction, ligase chain reaction).
- nucleic acid molecules encoding other polypeptide of the present invention could be isolated directly by using all or a substantial portion of the nucleic acid molecules of the present invention as DNA hybridization probes to screen cDNA or genomic libraries from any desired plant employing methodology well known to those skilled in the art. Methods for forming such libraries are well known in the art. Specific oligonucleotide probes based upon the nucleic acid molecules of the present invention can be designed and synthesized by methods known in the art.
- nucleic acid molecules can be used directly to synthesize DNA probes by methods known to the skilled artisan such as random primer DNA labeling, nick translation, or end-labeling techniques, or RNA probes using available in vitro transcription systems.
- specific primers can be designed and used to amplify a part or all of the sequences.
- the resulting amplification products can be labeled directly during amplification reactions or labeled after amplification reactions, and used as probes to isolate full-length cDNA or genomic DNAs under conditions of appropriate stringency.
- the nucleic acid molecules of interest can be amplified from nucleic acid samples using amplification techniques.
- the disclosed nucleic acid molecules may be used to define a pair of primers that can be used with the polymerase chain reaction (Mullis, et al, Cold Spring Harbor Symp. Quant. Biol. 57:263-273 (1986); Erlich et al, EP 50,424; EP 84,796, EP 258,017, EP 237,362; Mullis, EP 201,184; Mullis et al, US 4,683,202; Erlich, US 4,582,788; and Saiki, R.
- PCR and other in vitro amplification methods may also be useful, for example, to clone nucleotide sequences that encode for polypeptides to be expressed, to make nucleic acid molecules to use as probes for detecting the presence of the desired mRNA in samples, for nucleic acid sequencing, or for other purposes.
- two short segments of the nucleic acid molecules of the present invention may be used in polymerase chain reaction protocols to amplify longer nucleic acid molecules encoding homologues of a polypeptide of the present invention from DNA or RNA.
- the skilled artisan can follow the RACE protocol (Frohman et al., Proc. Natl. Acad. Sci. USA 85:8998 (1988), herein inco ⁇ orated by reference in its entirety) to generate cDNAs by using PCR to amplify copies of the region between a single point in the transcript and the 3 ' or 5' end.
- Primers oriented in the 3' and 5' directions can be designed from the nucleic acid molecules of the present invention.
- 3 'RACE or 5 'RACE systems Using commercially available 3 'RACE or 5 'RACE systems (Gibco BRL, Life Technologies, Gaithersburg, Maryland U.S.A.), specific 3' or 5' cDNA fragments can be isolated (Ohara et al., Proc. Natl. Acad. Sci. USA 86:5673 (1989); Loh et al., Science 243:217 (1989), both of which are herein inco ⁇ orated by reference in their entireties). Products generated by the 3' and 5' RACE procedures can be combined to generate full-length cDNAs (Frohman and Martin, Techniques ! : 165 (1989), herein incorporated by reference in its entirety).
- Nucleic acid molecules of interest may also be synthesized, either completely or in part, especially where it is desirable to provide plant-preferred sequences, by well-known techniques as described in the technical literature. See, e.g., Carruthers et al., Cold Spring Harbor Symp. Quant. Biol. 47:411-418 (1982), and Adams et al., J. Am. Chem. Soc. 105:661 (1983), both of which are herein inco ⁇ orated by reference in their entireties. Thus, all or a portion of the nucleic acid molecules of the present invention may be synthesized using codons preferred by a selected plant host. Plant-preferred codons may be determined, for example, from the codons used most frequently in the proteins expressed in a particular plant host species. Other modifications of the gene sequences may result in mutants having slightly altered activity
- nucleotide sequences encoding polypeptide of the present invention facilitates immunological screening of cDNA expression libraries.
- Synthetic polypeptides representing portions of the amino acid sequences of polypeptides of the present invention may be synthesized. These polypeptides can be used to immunize animals to produce polyclonal or monoclonal antibodies with specificity for polypeptides comprising the amino acid sequences. These antibodies can be then be used to screen cDNA expression libraries to isolate full-length cDNA clones of interest (Lemer, Adv. Immunol. 36: 1 (1984); Sambrook et al., Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, (1989)).
- Another aspect of the present invention relates to methods of making a DNA construct by obtaining a nucleic acid molecule comprising a nucleotide sequence encoding a polypeptide of the present invention, the amino acid sequence of which has at least 50% sequence identity to a polypeptide selected from the group consisting of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 6, SEQ ID NO: 8, SEQ ID NO: 10, SEQ ID NO: 12, SEQ ID NO: 14, SEQ ID NO: 16, SEQ ID NO: 18, SEQ ID NO: 20, SEQ ID NO: 22, SEQ ID NO: 24, SEQ ID NO: 26, SEQ ID NO: 28, SEQ ID NO: 30, SEQ ID NO: 32, SEQ ID NO: 34, SEQ ID NO: 36, SEQ ID NO: 38, SEQ ID NO: 40, SEQ ID NO: 42, SEQ ID NO: 44, SEQ ID NO: 46, SEQ ID NO: 48, SEQ ID NO: 50, SEQ ID NO: 52, SEQ ID NO: 54, SEQ ID NO:
- nucleic acid molecule comprising a nucleotide sequence or complement thereof, wherein the nucleotide sequence encodes a polypeptide having an amino acid sequence that has at least 50% sequence identity, preferably at least 60%, more preferably at least 70% sequence identity, even more preferably at least 80% or 90% sequence identity, and most preferably at least 95% to 98% sequence identity to a polypeptide selected from group consisting of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 6, SEQ ID NO: 8, SEQ ID NO: 10, SEQ ID NO: 12, SEQ ID NO: 14, SEQ ID NO: 16, SEQ ID NO: 18, SEQ ID NO: 20, SEQ ID NO: 22, SEQ ID NO: 24, SEQ ID NO: 26, SEQ ID NO: 28, SEQ ID NO: 30, SEQ ID NO: 32, SEQ ID NO: 34, SEQ ID NO: 36, SEQ ID NO: 38, SEQ ID NO: 40, SEQ ID NO: 42, SEQ ID NO: 44, SEQ ID NO:
- One method of the present invention is for obtaining a nucleic acid molecule encoding all or a substantial portion of the amino acid sequence of a polypeptide of the present invention comprising: (a) probing a cDNA or genomic library with a hybridization probe comprising a nucleotide sequence encoding all or a substantial portion of a polypeptide having an amino acid sequence set forth in any of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 6, SEQ ID NO: 8, SEQ ID NO: 10, SEQ ID NO: 12, SEQ ID NO: 14, SEQ ID NO: 16, SEQ ID NO: 18, SEQ ID NO: 20, SEQ ID NO: 22, SEQ ID NO: 24, SEQ ID NO: 26, SEQ ID NO: 28, SEQ ID NO: 30, SEQ ID NO: 32, SEQ ID NO: 34, SEQ ID NO: 36, SEQ ID NO: 38, SEQ ID NO: 40, SEQ ID NO: 42, SEQ ID NO: 44, SEQ ID NO: 46, SEQ ID NO: 48, SEQ
- Another method of the present invention for obtaining a nucleic acid molecule encoding all or a substantial portion of the amino acid sequence of a polypeptide of the present invention comprising: (a) synthesizing a first and a second oligonucleotide primers, wherein the sequences of the first and second oligonucleotide primers encode two different portions of a polypeptide having an amino acid sequence selected from the group consisting of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 6, SEQ ID NO: 8, SEQ ID NO: 10, SEQ ID NO: 12, SEQ ID NO: 14, SEQ ID NO: 16, SEQ ID NO: 18, SEQ ID NO: 20, SEQ ID NO: 22, SEQ ID NO: 24, SEQ ID NO: 26, SEQ ID NO: 28, SEQ ID NO: 30, SEQ ID NO: 32, SEQ ID NO: 34, SEQ ID NO: 36, SEQ ID NO: 38, SEQ ID NO: 40, SEQ ID NO: 42, SEQ ID NO: 44, SEQ ID NO
- nucleic acid molecules of the present invention may also be used as probes for genetically and physically mapping the genes that they are a part of, and as markers for traits linked to those genes. Such information may be useful in plant breeding in order to develop lines with desired phenotypes.
- the nucleic acid molecules of the present invention may be used as restriction fragment length polymo ⁇ hism (RFLP) markers. Southern blots (Maniatis et al., Molecular Cloning: A Laboratory Manual, Second Edition (1989) Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y., herein inco ⁇ orated by reference in its entirety) of restriction-digested plant genomic DNA may be probed with the nucleic acid fragments of the present invention.
- the resulting banding patterns may then be subjected to genetic analyses using computer programs such as MapMaker (Lander et al., Genomics 7:174-181 (1987), herein inco ⁇ orated by reference in its entirety) in order to construct a genetic map.
- the nucleic acid fragments of the present invention may be used to probe Southern blots containing restriction endonuclease-treated genomic DNAs of a set of individuals representing parent and progeny of a defined genetic cross. Segregation of the DNA polymo ⁇ hisms is noted and used to calculate the position of the nucleotide sequence of the present invention in the genetic map previously obtained using this population (Botstein et al, Am. J. Hum. Genet. 32:314-331 (1980), herein inco ⁇ orated by reference in its entirety).
- Nucleic acid probes derived from the nucleic acid molecules of the present invention may also be used for physical mapping (i.e., placement of sequences on physical maps; see Hoheisel et al, In: Nonmammalian Genomic Analysis: A Practical Guide, Academic press 1996, pp. 319- 346, and references cited therein).
- nucleic acid probes derived from the nucleic acid molecules of the present invention may be used in direct fluorescence in situ hybridization (FISH) mapping (Trask, Trends Genet. 7:149-154 (1991), herein incorporated by reference in its entirety).
- FISH direct fluorescence in situ hybridization
- current methods of FISH mapping favor use of large clones (several to several hundred KB; see Laan et al, Genome Res. 5:13-20 (1995), herein inco ⁇ orated by reference in its entirety)
- improvements in sensitivity may allow performance of FISH mapping using shorter probes.
- nucleic acid amplification-based methods of genetic and physical mapping may be carried out using the nucleotide molecules of the present invention.
- examples include allele-specific amplification (Kazazian et al., J. Lab. Clin. Med. 77:95-96 (1989), herein inco ⁇ orated by reference in its entirety), polymo ⁇ hism of PCR-amplified fragments (CAPS; Sheffield et al, Genomics 76:325-332 (1993), herein inco ⁇ orated by reference in its entirety), allele-specific ligation (Landegren et al, Science 241:1011-1080 (1988) herein inco ⁇ orated by reference in its entirety), nucleotide extension reactions (Sokolov et al., Nucleic Acid Res.
- Isolated nucleic acid molecules of the present invention may find use in the identification of loss of function mutant phenotypes of a plant, due to a mutation in one or more endogenous genes encoding polypeptides of the present invention. This can be accomplished either by using targeted gene disruption protocols or by identifying specific mutants for these genes contained in a population of plants carrying mutations in all possible genes (Ballinger and Benzer, Proc. Natl. Acad Sci USA 5(5:9402-9406 (1989); Koes et al, Proc. Natl. Acad. Sci. USA 92: 149-8153 (1995); Bensen et al, Plant Cell 7/75-84 (1995) all of which are incorporated herein by reference in their entirety).
- short segments of the nucleic acid molecules of the present invention may be used in polymerase chain reaction protocols in conjunction with a mutation tag sequence primer on DNAs prepared from a population of plants in which mutator transposons or some other mutation-causing DNA element has been introduced. The amplification of a specific DNA fragment with these primers indicates the insertion of the mutation tag element in or near the plant gene encoding polypeptide(s) of the present invention.
- the nucleic acid molecules of the present invention may be used as a hybridization probe against PCR amplification products generated from the mutation population using the mutation tag sequence primer in conjunction with an arbitrary genomic site primer, such as that for a restriction enzyme site-anchored synthetic adapter.
- Isolated nucleic acid molecules of the present invention can be mutated by a variety of methods well known in the art (Shortle D. et al., Annu. Rev. Genet. 15: 265, 1981; Itakura K. et al., Ann Rev. Biochem 53: 323, 1984; Botstein D. & Shortle D., Science 229: 1193, 1985; Smith M., Ann. Rev. Genet. 19: 423 1985; and Sambrook, et al, In: Molecular Cloning, A Laboratory Manual, 2nd Edition, Cold Spring Harbor Press, Cold Spring Harbor, New York 1989, herein incorporated by reference in its entirety).
- mutations can also be introduced by causing point mutation or site directed mutagenesis by using commercially available kits such as QuickChangeTM from Stratagene (11011 North Torrey Pines Road, La Jolla, CA 92037).
- site directed mutations can include but are not limited to, truncations, substitutions, additions, terminations, fusions of polypeptides or nucleic acid.
- a plant containing a mutation in the endogenous gene encoding polypeptides of the present invention can be identified and obtained.
- Such plant can also be obtained by in situ site directed mutagenesis. This mutant plant can then be used to determine or confirm the natural function of the polypeptides of the present invention disclosed herein.
- C-terminal region refers to the region of a peptide, polypeptide, or protein chain from the middle thereof to the end that carries the amino acid having a free carboxyl group.
- N-terminal region refers to the region of a peptide, polypeptide, or protein chain from the amino acid having a free amino group to the middle of the chain.
- Antisense Oligonucleotides or Polynucleotides of the Present Invention provides an antisense oligonucleotide or polynucleotide encoding an RNA molecule which hybridizes to at least a portion of an RNA transcript of an endogenous gene encoding a polypeptide of the present invention, wherein the RNA molecule hybridizes with the RNA transcript such that expression of the endogenous gene is altered.
- the present invention in another aspect, provides DNA construct wherein a promoter that functions in plant is operably linked to an antisense oligonucleotide or polynucleotide encoding an RNA molecule which hybridizes under stringent hybridization conditions to at least a portion of an RNA transcript of an endogenous gene encoding a polypeptide the amino acid sequence of which has at least 50% sequence identity to a member selected from the group consisting of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 6, SEQ ID NO: 8, SEQ ID NO: 10, SEQ ID NO: 12, SEQ ID NO: 14, SEQ ID NO: 16, SEQ ID NO: 18, SEQ ID NO: 20, SEQ ID NO: 22, SEQ ID NO: 24, SEQ ID NO: 26, SEQ ID NO: 28, SEQ ID NO: 30, SEQ ID NO: 32, SEQ ID NO: 34, SEQ ID NO: 36, SEQ ID NO: 38, SEQ ID NO: 40, SEQ ID NO: 42, SEQ ID NO: 44, SEQ ID NO
- the antisense oligonucleotide or polynucleotide can be full length or preferably has about six to about 100 nucleotides.
- the antisense oligonucleotide or polynucleotide hybridizes under stringent conditions to either at least a corresponding portion of one strand of a nucleotide sequence selected from the group consisting of SEQ ID NO: 1, SEQ ID NO: 3, SEQ ID NO: 5, SEQ ID NO: 7, SEQ ID NO: 9, SEQ ID NO: 11, SEQ ID NO: 13, SEQ ID NO: 15, SEQ ID NO: 17, SEQ ID NO: 19, SEQ ID NO: 21, SEQ ID NO: 23, SEQ ID NO: 25, SEQ ID NO: 27, SEQ ID NO: 29, SEQ ID NO: 31, SEQ ID NO: 33, SEQ ID NO: 35, SEQ ID NO: 37, SEQ ID NO: 39, SEQ ID NO: 41, SEQ ID NO: 43, SEQ ID NO: 45, SEQ ID NO: 47, SEQ ID NO:
- the antisense oligonucleotide or polynucleotide hybridizes under stringent conditions to a corresponding portion of the 5' non- coding region or 3' non-translated region of the RNA transcript.
- the antisense oligonucleotide or polynucleotide further comprises a sequence encoding a catalytic RNA or riboenzyme.
- the antisense oligonucleotides or polynucleotides of the present invention may find particular use in antisense technology to suppress endogenous gene expression to control sizes of organs in transgenic plants.
- a nucleic acid molecule derived from a nucleotide sequence selected from the group consisting of SEQ ID NO: 1, SEQ ID NO: 3, SEQ ID NO: 5, SEQ ID NO: 7, SEQ ID NO: 9, SEQ ID NO: 11, SEQ ID NO: 13, SEQ ID NO: 15, SEQ ID NO: 17, SEQ ID NO: 19, SEQ ID NO: 21, SEQ ID NO: 23, SEQ ID NO: 25, SEQ ID NO: 27, SEQ ID NO: 29, SEQ ID NO: 31, SEQ ID NO: 33, SEQ ID NO: 35, SEQ ID NO: 37, SEQ ID NO: 39, SEQ ID NO: 41, SEQ ID NO: 43, SEQ ID NO: 45, SEQ ID NO: 47, SEQ ID NO: 49, SEQ ID NO: 51, SEQ ID NO: 53, SEQ ID NO: 66, SEQ ID NO: 68, and SEQ ID NO: 70 is cloned and operably linked to a promoter such that the antisense strand of RNA will be
- antisense RNA inhibits gene expression by preventing the accumulation of mRNA that encodes the enzyme of interest (see, e.g., Sheehy et al, Proc. Nat. Acad. Sci. USA 55:8805-8809 (1988), and U.S. Patent No. 4,801,340; both of which are herein incorporated by reference in their entireties).
- the antisense oligonucleotide or polynucleotide to be introduced generally will be substantially identical to at least a portion of the endogenous gene or genes of the present invention to be repressed.
- the sequence however, needs not to be perfectly identical to inhibit or suppress expression of the endogenous gene or genes of the present invention.
- the introduced sequence also needs not be full length relative to either the primary transcription product or fully processed mRNA. Generally, higher homology can be used to compensate for the use of a shorter sequence. Furthermore, the introduced sequence need not have the same intron or exon pattern, and homology of non-coding segments may he equally effective. Normally, a sequence of between about 6 nucleotides and about full length nucleotides should be used, though a sequence of at least about 500 to about 1700 nucleotides is preferred, a sequence of at least about 200 nucleotides is more preferred, and a sequence of about 6 to about 100 nucleotides is especially preferred.
- the antisense oligonucleotide or polynucleotide is substantially complementary to at least a corresponding portion of one strand of a nucleotide sequence selected from the group consisting of SEQ ID NO: 1, SEQ ID NO: 3, SEQ ID NO: 5, SEQ ID NO: 7, SEQ ID NO: 9, SEQ ID NO: 11, SEQ ID NO: 13, SEQ ID NO: 15, SEQ ID NO: 17, SEQ ID NO: 19, SEQ ID NO: 21, SEQ ID NO: 23, SEQ ID NO: 25, SEQ ID NO: 27, SEQ ID NO: 29, SEQ ID NO: 31, SEQ ID NO: 33, SEQ ID NO: 35, SEQ ID NO: 37, SEQ ID NO: 39, SEQ ID NO: 41, SEQ ID NO: 43, SEQ ID NO: 45, SEQ ID NO: 47, SEQ ID NO: 49, SEQ ID NO: 51, SEQ ID NO: 53, SEQ ID NO: 66, SEQ ID NO: 68, SEQ ID NO: 70, or
- the antisense oligonucleotide or polynucleotide is substantially complementary to a corresponding portion of the 5' non-coding portion or 3' non-coding portion of one strand of a DNA molecule which has substantial sequence identity to a sequence selected from the group consisting of SEQ ID NO: 1, SEQ ID NO: 3, SEQ ID NO: 5, SEQ ID NO: 7, SEQ ID NO: 9, SEQ ID NO: 11, SEQ ID NO: 13, SEQ ID NO: 15, SEQ ID NO: 17, SEQ ID NO: 19, SEQ ID NO: 21, SEQ ID NO: 23, SEQ ID NO: 25, SEQ ID NO: 27, SEQ ID NO: 29, SEQ ID NO: 31, SEQ ID NO: 33, SEQ ID NO: 35, SEQ ID NO: 37, SEQ ID NO: 39, SEQ ID NO: 41, SEQ ID NO: 43, SEQ ID NO: 45, SEQ ID NO: 47, SEQ ID NO: 49, SEQ ID NO: 51, SEQ ID NO: 53, SEQ ID NO: 66,
- the antisense oligonucleotides or polynucleotides of the present invention may further comprise a nucleotide sequence encoding a catalytic RNA molecule or a ribozyme. It is known to a skilled person in the art that catalytic RNA molecules or ribozymes can also be used to inhibit expression of genes of the present invention. It is possible to design ribozymes that specifically pair with virtually any target RNA and cleave the phosphodiester backbone at a specific location, thereby functionally inactivating the target RNA. In carrying out this cleavage, the ribozyme is not itself altered, and is thus capable of recycling and cleaving other molecules, making it a true enzyme. The inclusion of ribozyme sequences within antisense RNAs confers RNA-cleaving activity upon them, thereby increasing the activity of the recombinant DNA constructs.
- RNAs A number of classes of ribozymes have been identified.
- One class of ribozymes is derived from a number of small circular RNAs, which are capable of self-cleavage and replication in plants.
- the RNAs replicate either alone (viroid RNAs) or with a helper virus (satellite RNAs). Examples include RNAs from avocado sunblotch viroid and the satellite RNAs from tobacco ringspot virus, lucerne transient streak virus, velvet tobacco mottle virus, Solanum nodiflorum mottle virus and subterranean clover mottle virus.
- the design and use of target RNA- specific ribozymes is described in Haseloff et al. Nature 334:585-591 (1988), herein incorporated by reference in its entirety.
- the present invention also provides antibodies that specifically bind to the polypeptides of the present invention and recombinant DNA constructs that comprise nucleic acid molecules of the present invention.
- transgenic plant means a plant that contains an exogenous nucleic acid, which can be derived from the same plant species or from a different species.
- exogenous it is meant that a nucleic acid molecule originates from outside the plant to which the nucleic acid molecule is introduced.
- An exogenous nucleic acid molecule can have a naturally occurring or non-naturally occurring nucleotide sequence.
- an exogenous nucleic acid molecule can be a heterologous nucleic acid molecule derived from a different plant species than the plant into which the nucleic acid molecule is introduced or can be a nucleic acid molecule derived from the same plant species as the plant into which it is introduced.
- the term "genome” as it applies to plant cells encompasses not only chromosomal DNA found within the nucleus, but organelle DNA found within subcellular components of the cell. DNAs of the present invention introduced into plant cells can therefore be either chromosomally integrated or organelle-localized.
- the term "genome” as it applies to bacteria encompasses both the chromosome and plasmids within a bacterial host cell. Encoding DNAs of the present invention introduced into bacterial host cells can therefore be either chromosomally integrated or plasmid-localized.
- Exogenous nucleic acid molecules may be transferred into a plant cell by the use of a recombinant DNA construct (or vector) designed for such a pu ⁇ ose.
- the present invention also provides a plant recombinant DNA construct (or vector) for producing transgenic plants, wherein the plant recombinant DNA construct (or vector) comprises a sense oligonucleotide or polynucleotide or an antisense oligonucleotide or polynucleotide.
- Methods that are well known to those skilled in the art may be used to prepare the plant recombinant DNA construct (or vector) of the present invention. These methods include in vitro recombinant DNA techniques, synthetic techniques, and in vivo genetic recombination.
- a plant recombinant DNA construct (or vector) of the present invention contains a sense oligonucleotide or polynucleotide or an antisense oligonucleotide or polynucleotide and operably linked regulatory sequences or control elements.
- exemplary regulatory sequences include but are not limited to promoters, translation leader sequences, introns, 3' non-translated sequences.
- the promoters can be constitutive, inducible, or preferably tissue-specific promoters.
- a plant recombinant DNA construct (vector) of the present invention will typically comprise a selectable marker that confers a selectable phenotype on plant cells.
- Selectable markers may also be used to select for plants or plant cells that contain the exogenous nucleic acid molecules of the present invention.
- the marker may encode biocide resistance, antibiotic resistance (e.g., kanamycin, G418 bleomycin, hygromycin, etc.), or herbicide resistance (e.g., glyphosate, etc.).
- selectable markers include, but are not limited to, a neo gene (Potrykus et al, Mol. Gen. Genet.
- a plant recombinant DNA construct (vector) of the present invention may also include a screenable marker. Screenable markers may be used to monitor expression. Exemplary screenable markers include a ⁇ -glucuronidase or uidA gene (GUS) which encodes an enzyme for which various chromogenic substrates are known (Jefferson, Plant Mol. Biol, Rep. 5:387-405 (1987); Jefferson et al, EMBO J.
- GUS ⁇ -glucuronidase or uidA gene
- selectable or screenable marker genes are also genes which encode a secretable marker whose secretion can be detected as a means of identifying or selecting for transformed cells. Examples include markers that encode a secretable antigen that can be identified by antibody interaction, or even secretable enzymes which can be detected catalytically.
- Secretable proteins fall into a number of classes, including small, diffusible proteins detectable, e.g., by ELISA, small active enzymes detectable in extracellular solution (e.g., ⁇ -amylase, ⁇ -lactamase, phosphinothricin transferase), or proteins which are inserted or trapped in the cell wall (such as proteins which include a leader sequence such as that found in the expression unit of extension or tobacco PR-S).
- small active enzymes detectable in extracellular solution e.g., ⁇ -amylase, ⁇ -lactamase, phosphinothricin transferase
- proteins which are inserted or trapped in the cell wall such as proteins which include a leader sequence such as that found in the expression unit of extension or tobacco PR-S.
- Other possible selectable and/or screenable marker genes will be apparent to those of skill in the art.
- reporter gene In addition to a selectable marker, it may be desirous to use a reporter gene. In some instances a reporter gene may be used with or without a selectable marker. Reporter genes are genes which are typically not present in the recipient organism or tissue and typically encode for proteins resulting in some phenotypic change or enzymatic property. Examples of such genes are provided in K. Wising et al. Ann. Rev. Genetics, 22, 421 (1988), which is inco ⁇ orated herein by reference. Preferred reporter genes include the beta-glucuronidase (GUS) of the uidA locus of E. coli, the chloramphenicol acetyl transferase gene from Tn9 of E.
- GUS beta-glucuronidase
- an assay for detecting reporter gene expression may then be performed at a suitable time after said gene has been introduced into recipient cells.
- a preferred such assay entails the use of the gene encoding beta-glucuronidase (GUS) of the uidA locus of E. coli as described by Jefferson et al., (Biochem. Soc. Trans. 15, 17-19 (1987)) to identify transformed cells.
- GUS beta-glucuronidase
- the various components of the construct or fragments thereof will normally be inserted into a convenient cloning vector, e.g., a plasmid that is capable of replication in a bacterial host, e.g., E. coli.
- a convenient cloning vector e.g., a plasmid that is capable of replication in a bacterial host, e.g., E. coli.
- the cloning vector with the desired insert may be isolated and subjected to further manipulation, such as restriction digestion, insertion of new fragments or nucleotides, ligation, deletion, mutation, resection, etc. so as to tailor the components of the desired sequence.
- a plant recombinant DNA construct (vector) of the present invention may also include a chloroplast transit peptide, in order to target the polypeptide of the present invention to the plastid.
- plastid means the class of plant cell organelles that includes amyloplasts, chloroplasts, chromoplasts, elaioplasts, eoplasts, etioplasts, leucoplasts, and proplastids. These organelles are self-replicating, and contain what is commonly referred to as the "chloroplast genome," a circular DNA molecule that ranges in size from about 120 to about 217 kb, depending upon the plant species, and which usually contains an inverted repeat region.
- chloroplast transit peptide examples include the small subunit of ribulose- 1 ,5-biphosphate carboxylase (ssRUBISCO, SSU), 5-enolpyruvateshikimate-3-phosphate synthase (EPSPS), ferredoxin, ferredoxin oxidoreductase, the light-harvesting-complex protein I and protein II, and thioredoxin F.
- chloroplast polypeptides include the small subunit of ribulose- 1 ,5-biphosphate carboxylase (ssRUBISCO, SSU), 5-enolpyruvateshikimate-3-phosphate synthase (EPSPS), ferredoxin, ferredoxin oxidoreductase, the light-harvesting-complex protein I and protein II, and thioredoxin F.
- non-plastid polypeptides may be targeted to the chloroplast by use of polypeptide fusions with a CTP and that a CTP sequence is sufficient to target a polypeptide to the plastid.
- a CTP sequence is sufficient to target a polypeptide to the plastid.
- Those skilled in the art will also recognize that various other recombinant DNA constructs can be made that utilize the functionality of a particular plastid transit peptide to import the enzyme into the plant cell plastid depending on the promoter tissue specificity.
- the present invention also provides a transgenic plant comprising in its genome an exogenous nucleic acid molecule which comprises: (A) a 5' non-coding sequence which functions in the cell to cause the production of an RNA molecule; which is operably linked to (B) an antisense oligonucleotide or polynucleotide or a sense oligonucleotide or polynucleotide of this invention; which is operably linked to (C) a 3' non-translated sequence that functions in said cell to cause termination of transcription.
- Transgenic plants of the present invention preferably have inco ⁇ orated into their genome or transformed into their chloroplast or plastid genomes an exogenous nucleic acid molecule (or "transgene") that comprises a sense oligonucleotide or polynucleotide or an antisense oligonucleotide or polynucleotide.
- Transgenic plants are also meant to comprise progeny (descendant, offspring, etc.) of any generation of such a transgenic plant.
- a seed of any generation of all such transgenic plants wherein said seed comprises a sense oligonucleotide or polynucleotide or an antisense oligonucleotide or polynucleotide of the present invention is also an important aspect of the invention.
- the DNA constructs of the present invention may be introduced into the genome of a desired plant host by a variety of conventional transformation techniques, which are well known to those skilled in the art.
- Preferred methods of transformation of plant cells or tissues are the Agrobacterium mediated transformation method and the biolistics or particle-gun mediated transformation method.
- Suitable plant transformation vectors for the purpose of Agrobacterium mediated transformation include those derived from a Ti plasmid of Agrobacterium tumefaciens, as well as those disclosed, e.g., by Herrera-Estrella et al., Nature 303:209 (1983); Bevan, Nucleic Acids Res.
- a plasmid expression vector suitable for the introduction of an antisense oligonucleotide or polynucleotide or a sense oligonucleotide or polynucleotide in monocots using electroporation or particle-gun mediated transformation is composed of the following: a promoter that is inducible or constitutive or tissue-specific; an intron that provides a splice site to facilitate expression of the gene, such as the Hsp70 intron (U.S. Patent Number 5,859,347); and a 3' polyadenylation sequence such as the nopaline synthase 3' sequence (NOS 3'; Fraley et al., Proc. Natl. Acad. Sci. USA 80: 4803-4807(1983)).
- This expression cassette may be assembled on high copy replicons suitable for the production of large quantities of DNA.
- US Patent No. 6,147,278 herein inco ⁇ orated by reference in its entirety, and contains a gene encoding an EPSPS enzyme conferring glyphosate resistance (denominated aroA; CP4), which is an excellent selection marker gene for many plants.
- the gene is fused to the Arabidopsis EPSPS chloroplast transit peptide (CTP2) and expressed from the FMV promoter as described therein.
- the cells (or protoplasts) can be cultured to regenerate into whole plants.
- Such regeneration techniques rely on manipulation of certain phytohormones in a tissue culture growth medium, typically relying on a biocide and/or herbicide marker which has been introduced together with the desired nucleotide sequences.
- Choice of methodology for the regeneration step is not critical, with suitable protocols being available for hosts from Leguminosae (alfalfa, soybean, clover, etc.), Umbelliferae (carrot, celery, parsnip), Cruciferae (cabbage, radish, canola/rapeseed, etc.), Cucurbitaceae (melons and cucumber), Gramineae (wheat, barley, rice, maize, etc.), Solanaceae (potato, tobacco, tomato, peppers), various floral crops, such as sunflower, and nut-bearing trees, such as almonds, cashews, walnuts, and pecans. See, for example, Ammirato et al., Handbook of Plant Cell Culture - Crop Species.
- a transgenic plant formed using Agrobacterium transformation methods typically contains a single exogenous gene on one chromosome. Such transgenic plants can be referred to as being heterozygous for the added exogenous gene.
- transgenic plant that is homozygous for the added exogenous gene; i.e., a transgenic plant that contains two added exogenous genes, one gene at the same locus on each chromosome of a chromosome pair.
- a homozygous transgenic plant can be obtained by sexually mating (selfing) an independent segregant transgenic plant that contains a single exogenous gene, germinating some of the seed produced and analyzing the resulting plants produced for the exogenous gene of interest.
- transgenic plants containing the exogenous nucleic acid molecule that encodes a polypeptide of interest are well known in the art.
- the regenerated plants are self-pollinated to provide homozygous transgenic plants, as discussed above. Otherwise, pollen obtained from the regenerated plants is crossed to seed-grown plants of agronomically important lines. Conversely, pollen from plants of these important lines is used to pollinate regenerated plants.
- a transgenic plant of the present invention containing desired polypeptides of the present invention are cultivated using methods well known to one skilled in the art.
- Plants that can be made to have larger organ size by practice of the present invention include, but are not limited to, Ac ⁇ ci ⁇ , alfalfa, aneth, apple, apricot, artichoke, arugula, asparagus, avocado, banana, barley, beans, beet, blackberry, blueberry, broccoli, brussels sprouts, cabbage, canola, cantaloupe, carrot, cassava, cauliflower, celery, cherry, cilantro, citrus, Clementines, coffee, corn, cotton, cucumber, Douglas fir, eggplant, endive, escarole, eucalyptus, fennel, figs, gourd, grape, grapefruit, honey dew, jicama, kiwifruit, lettuce, leeks, lemon, lime, loblolly pine, mango, melon, mushroom, nut, oat, okra, onion, orange, an ornamental plant, papaya, parsley, pea, peach, peanut, pear, pepper, persimmon, pine
- the present invention also provides cells of the transgenic plants of the present invention which could be used for regenerating a plant or any organ of plant with present invention.
- the present invention also further provides a method for altering a specific organ or organs in a plant to generate smaller plant or plant organ, the method comprising the steps of: a) introducing into the genome of the plant an exogenous nucleic acid molecule comprising in the 5' to 3' direction i) a promoter that functions in the cells of said plant, said promoter operably linked to; ii) an antisense oligonucleotide or polynucleotide or a sense oligonucleotide or polynucleotide of the present invention, said antisense oligonucleotide or polynucleotide operably linked to; iii) a 3' non-translated nucleotide sequence that functions in said cells of said plant to cause transcriptional termination; b) obtaining transformed plant cells containing the exogenous nucleic acid molecule of step (a); and c) regenerating from said transformed plant cells a transformed plant in which the expression of an endogenous gene of the
- Arabidopsis thaliana var Columbia seeds were obtained from Lehle seeds (LEHLE SEEDS 1102 South Industrial Blvd., Suite D, Round Rock TX 78681 USA). For growing seeds into plants, 2.5 inch pots are prepared with soil covered with bridal veil or a mesh screen, making sure that the soil is not packed too tightly and the mesh is in contact with the soil surface (this ensures that the germinating seedlings will be able to get through the mesh. Seeds are sown and covered with a germination dome. Seeds are vernalized for 3-4 days. Plants are grown under conditions of 16 hours light / 8 hours dark at 20-22° C, 70% humidity.
- MF16 5'SAG13 and MF17 3'SAG13 were designed based on the SAG 13 sequence information (AF192276) from Genbank and custom synthesized by Gibco BRL, Life Technologies, Gaithersburg, Maryland U.S. A.
- MF16 5'SAG13 is ATA TTT AAC AAG CCA TGG CAA AGG A
- MF17 3'SAG13 is ATA TGT GTT TGA ATT CAT AGT CTT GAA identified as SEQ ID NO: 55 and SEQ ID NO: 56 respectively in the sequence list, which annealed on SAG 13 gene to introduce Nco 1 site at 5 'end and Eco RI site at 3 'end of the gene.
- PCR was then performed to amplify the S.4G-73 cDNA using the above prepared cDNA as the template, and MF16 5'SAG13 and MF17 3'SAG13 as the primers.
- the thermal cycling conditions were as follows: 94°C, 40 second, followed 30 cycles of 94°C, 25 seconds; 55°C, 30 seconds and 68°C, 2 minutes 30 seconds ( All reagents and equipment for PCR can be procured from Applied Biosystems, 850 Lincoln Center Drive, Foster City, CA 94404, USA).
- the amplified cDNA was purified by gel-electrophoresis to obtain the gene identified as SEQ ID NO: 1.
- RNA is isolated from appropriate crop and other desired plant species by pooling tissues of different developmental stages of all vegetative and reproductive organs. Sequences can be cloned out from total RNA by methodology shown in above paragraph.
- DNA is isolated from the desired microorganism. Isolation of DNA from microorganism is well known in the art (Sambrook, et al, In: Molecular Cloning, A Laboratory Manual, 2nd Edition, Cold Spring Harbor Press, Cold Spring Harbor, New York 1989, herein inco ⁇ orated by reference in its entirety). This DNA along with oligonucleotide PCR primers can be used in a polymerase chain reaction by any one skilled in the art to isolate genes of the invention.
- mutations can also be introduced by causing a point mutation or site directed mutagenesis in nucleotide SEQ ID NO: 3 by using commercially available kits such as QuickChangeTM Site Directed Mutagenesis Kit from Stratagene (11011 North Torrey Pines Road, La Jolla, CA 92037) to result in a peptide with about 90% of the N-terminal of SEQ ID NO: 4 by altering a nucleotide sequence to encode a stop codon at amino acid position 241 + 3, or to result in a peptide with about 80% of the N- terminal of SEQ ID NO: 4 by altering a nucleotide sequence to encode a stop codon at amino acid position 214 + 3, or to result in about 70% of the N-terminal of SEQ ID NO: 4 by altering a nucleotide sequence to encode a stop codon at amino acid position 188 ⁇ 3, or to result in about 60%) of the N-terminal of SEQ ID NO: 4 by altering a nucleotide
- mutations can also be introduced in situ by causing point mutation or site directed mutagenesis in nucleotide SEQ ID NO: 3 by using in situ mutagenesis technology provided by Valigen Inc. (Newtown PA. USA) (US Patent Number 6,211,351; US Patent Number 6,271,360, WO 01/24615 Al, and WO 01/25460 A2, herein incorporated by reference in its entirety) to result in a peptide with about 90% of the N-terminal of SEQ ID NO: 4 by altering a nucleotide sequence to encode a stop codon at amino acid position 241 ⁇ 3, or to result in a peptide with about 80% of the N-terminal of SEQ ID NO: 4 by altering a nucleotide sequence to encode a stop codon at amino acid position 214 + 3, or to result in about 70% of the N-terminal of SEQ ID NO: 4 by altering a nucleotide sequence to encode a stop codon at amino acid position 188 + 3, or to result in about 60% of
- DNA segments that provide replication function and antibiotic selection in bacterial cells for example, an E. coli origin of replication such as or/322, a broad host range origin of replication such as orN or oriRi, and a coding region for a selectable marker such as Spc/Str that encodes for Tn7 aminoglycoside adenyltransferase (aadA) conferring resistance to spectinomycin or streptomycin, or a gentamicin (Gm, Gent) selectable marker.
- the host bacterial strain is Agrobacterium tumefaciens ABI or LBA4404.
- the genetic elements of the DNA constructs are assembled to have in operable linkage a promoter that functions in plants. Additionally an antibiotic or herbicide marker cassette, an epitope tag (for example Flag® peptide catalog number F-3290, SIGMA, P.O. Box 14508 St. Louis, MO 63178 USA) at the 3' termination region of gene of interest is included in the DNA construct.
- the multiple cloning site in this DNA construct encodes Bglll, Ncol, EcoRI, Sail, and Xhol.
- the epitope tag region was encoded with Sail and Xhol restriction sites for optional removal of the epitope tag.
- the Ncol site encodes a Kozak sequence for efficient translation of the protein products.
- DNA constructs used in the method of the current invention comprise any promoter known to function to cause the transcription in plant cells and any antibiotic tolerance encoding polynucleotide sequence known to confer antibiotic tolerance to plant cells.
- the antibiotic tolerance polynucleotide sequences include, but are not limited to polynucleotide sequences encoding for proteins involved in antibiotic tolerance to kanamycin, neomycin, hygromycin, and other antibiotics known in the art.
- Antibiotic tolerance gene in such a vector can be replaced by herbicide tolerance encoding for 5-enolpyruvylshikimate-3 -phosphate synthase (EPSPS, described in U.S.
- EPSPS 5-enolpyruvylshikimate-3 -phosphate synthase
- Patent Number 4,810,648 phytoene desaturase (crtl (Misawa et al, (1993) Plant Journal 4:833-840, and (1994) Plant Jour 6:481-489) for tolerance to norflurazon, acetohydroxy acid synthase (AHAS, Sathasiivan et al. (1990) Nucl. Acids Res. 18:2188-2193) and the bar gene for tolerance to glufosinate (DeBlock, et al. (1987) E R ⁇ J. 6:2513-2519.
- Herbicides for which transgenic plant tolerance has been demonstrated and the method of the present invention can be applied include, but are not limited to: glyphosate, glufosinate, sulfonylureas, imidazolinones, bromoxynil, delapon, cyclohezanedione, protopo ⁇ hyrionogen oxidase inhibitors, and isoxaslutole herbicides.
- P- ⁇ 35S promoter U.S. Patent No.5,539,142, 5,196,525, 5,322,938 and 5,164,316 herein incorporated by reference in its entirety.
- P-E35S promoter can be replaced by P-CaMV.35S promoter (U.S. Patent No. 5,858,742, herein inco ⁇ orated by reference in its entirety), or by enhanced P-CaMV.35S from Cauliflower mosaic virus containing a duplication of the -90-300 region as described in U. S. Patent No.
- Figwort mosaic virus promoter P-FMV, as described in U. S. Patent No. 5,378,619, herein inco ⁇ orated by reference in its entirety; or the P-AtEFla (P-AtEFl or EFla) a promoter region from the Arabidopsis thaliana elongation factor gene la; the GboxlO and Gboxl lmotif (Fumiharu et al., (1999) Plant J. 18:443-448); or the DC3 promoter region from carrot (Seffens et al., Develop. Genet. 11:65-76); or the TP12 promoter (GenBank accession no.
- DNA molecules encoding plastid transit peptides for example, the Arabidopsis EPSPS chloroplast transit peptide, At.CTP2 as described in U. S. Patent No. 5,633,435, herein inco ⁇ orated by reference in its entirety.
- the method of the present invention enables one of skill in the art of plant molecular biology to design and assemble plant expression cassettes that contain promoters of known and unknown function.
- the genetic elements of the DNA construct further comprise 5' leader polynucleotides for example, the Hsp70 non-translated leader sequence from Petunia hybrida as described in U. S. Patent No. 5,362,865, herein inco ⁇ orated by reference in its entirety.
- the genetic elements further comprise herbicide tolerance genes that include, but are not limited to, for example, the aroA:CP4 coding region for EPSPS glyphosate resistant enzyme isolated from Agrobacterium tumefaciens (AGRTU) strain CP4 as described in U. S. Patent No. 5,633,435, herein incorporated by reference in its entirety.
- the genetic elements of the DNA construct further comprise 3' termination regions that include, but are not limited to, the E9 3' termination region of the pea RbcS gene that functions as a polyadenylation signal; the nos is the 3' end of the nopaline synthase gene that functions as a polyadenylation signal.
- the genetic elements of the DNA construct further comprise the right border (RB) and left borders (LB) of the Ti plasmid of Agrobacterium tumefaciens octopine and nopaline strains.
- construct pMON575221 was transformed in Arabidopsis plants by Agrobacterium mediated transformation procedure.
- Amplified and purified product SEQ ID NO: 1 can also be cloned in antisense orientation in appropriate cloning sites of vector pMON23435 to express the opposite strand of SEQ ID NO:l by one skilled in the art.
- EXAMPLE 4 Cloning of DNA molecules of the present invention: This example illustrates how all other DNA molecules as shown in table from example 9 are isolated from different plant species by designing appropriate PCR primers based on the DNA sequence information provided in the table of Example 8. For isolating these DNA molecules one skilled in the art will isolate total RNA from a crop or other desired plant species by pooling tissues of different developmental stages of all vegetative and reproductive organs. DNA molecules are cloned out from total RNA by methodology shown in Example 2.
- Isolated DNA molecule sequences can be cloned at appropriate cloning sites in sense or antisense orientation of a plant expression vector shown in Example 3 or a similar vector capable of ectopically expressing the gene of interest of the present invention in sense or antisense orientation as known in the art.
- This example illustrates how Agrobacterium cells are transformed and how transformed cells are cultured. Transformation ; 1. Electroporate 2 ⁇ l of DNA construct into 20 ⁇ l of ABI competent cells;
- ABI Cell Culture 1. Pick 3 colonies per ABI plate and grow each in 4ml LB media containing Spectinomycin (75 ⁇ g/ml), Kanamycin (50 ⁇ g/ml), Chloramphenicol (25 ⁇ g/ml);
- Glycerol Stocks. & DNA Preps 1. Make three 1 ml ABI glycerol stocks per 4 ml culture, using 500 ⁇ l of culture and 500 ⁇ l of 40% glycerol. Freeze and store at -80°C. 2. Miniprep remaining culture (about 2.5ml), using a Qiagen miniprep kit and protocol (Qiagen Genomics, Inc., Seattle, WA), ensuring add PB buffer wash step and EB buffer (lOmM Tris- Cl, pH 8.5) to 70°C before eluting DNA from column. The resulting volume per miniprep sample should be 50 ⁇ l.
- Arabidopsis plants may be transformed by any one of many available methods.
- Arabidopsis plants may be transformed using in planta transformation method by vacuum infiltration (see, Bechtold et al, In planta Agrobacterium mediated gene transfer by infiltration of adult Arabidopsis thaliana plants. CR Acad. Sci. Paris Sciences de la vie/life sciences 316: 1194-1199 (1993), herein inco ⁇ orated by reference in its entirety). Plants can be grown as described in Example 1.
- Asrobacterium Preparation (Small scale and Large scale cultures ' ): Agrobacterium strain ABI is streaked onto an LB plate containing Spectinomycin 100 mg/L, Streptomycin 100 mg/L, Chloramphenicol 25mg/L, and Kanamycin 50mg/L (denoted SSCK). Two days prior to infiltration, a loop of Agrobacterium is placed into a tube containing 10 mis LB/SSCK and put on a shaker in the dark at 28°C to grow overnight. The following day, the Agrobacterium is diluted 1:50 in 400 mis of bacterial grown medium such as SSCK and put on a shaker at 28°C to grow for 16-20 hours. Infiltration
- a very small amount of seed can be first plated out to check for contamination. If there is contamination, re-sterilized seeds for ⁇ 4 more hours and check for contamination again.
- the second sterilization is usually not necessary, but sometimes the seed harbors a fungal contaminant and repeat sterilizations are needed. (The sterilization duration generally is shorter than 16 hours because of significantly decreased germination rates starting at 24 hr. sterilization duration).
- Example 7 Following subsections of Example 7 are used for describing phenotypic changes after transformation and growth of Arabidopsis plants as described in Example 6.
- Three different events obtained from transformation of pMON57521 (Figure 2) into Arabidopsis thaliana ecotype Columbia were selected and grown side by side with wild type plants. Growth conditions were 16 hr light, 8 hr night, 21 degrees Centigrade and 70% relative humidity. Plants were observed at all growth stages and photographed using an Olympus camera C-2500 L (Olympus America Inc., 2 Corporate Center Drive, Melville, NY 11747) as described by the manufacturer.
- Olympus America Inc. 2 Corporate Center Drive, Melville, NY 11747
- Plants transformed with Construct pMON57521 were shown to exhibit 2 to 3 times larger flower and floral organs such as stamen and pistils when compared to the wild type, non transformed plants of the same species. Transgenic plants were also observed to have at least 2 times more lateral roots and 2 to 3 times thicker stem size when compared to the lateral roots and stems of wild type, non transformed plants of the same species. Approximately 2 fold increased trichome numbers and distribution were also observed. Over expression of the polypeptide encoded by the sequence in the pMON57521 construct of the present invention in the transformed plants, yielded plants that exhibited a 100 % increase in individual seed size and weight.
- Seeds from transformed plants exhibit increased seed size that was approximately twice as large as the seeds from wild type plants as shown in following table. Image sizes were determined by the Pro or Lucis software ( Optronics, 175 Cremona Drive, Goleta, CA 93117) based on the image pixel value of wild type and transformed plants' seed under the same resolution. Plant lines 8752-1, 8752-2, 8752-6, and 8752-7 in Table 1 correspond to different transgenic plant lines produced by transforming wild Arabidopsis plants with pMON57521.
- Seed Weight The seed from plants containing pMON57521 lines was also found to be heavier than WT seed. Seed weight was measured from 2 lines produced from event 8752 (8752-1 and 8752-6) and 2 lines from event 8748 (8748-2 and 8748-7) and compared with wild type. Shown below is a representative seed weight analysis. For each line, 3 replications were measured (i.e. 50 seeds were counted three times and measured each time). This analysis was repeated with different seed counts (eg. 100 seed or 150 seed) and was found to be highly reproducible. For example, weight per 100 seed is similarly high ⁇ 0.0025 gm. for wild type and 0.0046 gm. for seeds from plants transformed with sequence of the present invention.
- Plant lines 8752-1, 8752-6, 8748-2, and 8748- 7 in Table 2 correspond to different transgenic plant lines produced by transforming wild Arabidopsis plants with pMON57521. Table 2: Seed Weight
- (C) Seed Number Lines over expressing sequence of the present invention were found to have fewer seed per silique as compared to wild type plants. The average number of seed /silique of equivalent maturity was 34 for transgenic plants of the present invention as compared to 52 for wild type plants. The analysis was repeated three times with highly reproducible results within and between events.
- Seed Yield There was an increase of > 20% seed yield in transgenic plants of the present invention. Seed yield when measured in terms of weight of total seeds per plant.
- silique number at growth stage 6.5 was estimated for both WT and transgenics and total silique number is shown in the following table. This indicates again that silique number was at least 28% greater in lines over-expressing the polypeptide molecules corresponding SEQ ID NO: 2 (transformed lines) as compared to wild type lines at the same growth stage. As the plant proceeds to senescence, the relative increase appears to stay consistent and together with the increase in seed weight in the transgenic plant, contributes to the overall increase in seed yield per plant. Growth stage estimation was based on "Growth stage- based phenotypic analysis of Arabidopsis: A model for high throughput functional genomics in plants. Plant Cell. 13(7):1499, 2001"
- WT wild type plant and Transformed line is transgenic plant line expressing polypeptide molecules corresponding to SEQ ID. NO: 2.
- This example describes increase of seeds in transgenic soybean plants expressing polypeptide molecule corresponding to SEQ ID NO: 2. Soybean plants were transformed with pMON73955 ( Figure 7) to constitutively express
- Soybean transformation is performed essentially as described in WO 00/42207, herein inco ⁇ orated by reference in its entirety. RI seed from 10 events out of 44 events were advanced in Puerto Rico (PR) based on the gene copy number in plants. Soybean transformation was performed essentially as described in WO 00/42207, herein inco ⁇ orated by reference in its entirety. Preliminary data indicates that 1 of the 10 events in PR showed a phenotype similar to that seen for the Arabidopsis over-expression of SEQ ID NO: 2 (pMON 57521 Figure 2) i.e. more pods and more branches as well as short, fat pods with fewer seed.
- Table 7 R2 seed weights of individual lines from the large-seeded transgenic event "A expressing SEQ ID NO: 2.
- This example describes over expression of the protein in bacterial cells for purification of the protein so as to screen for the activity of polypeptide molecules.
- A Cloning of the nucleotide molecules of the present invention for expression of corresponding peptides.
- pMON57521 Figure 2
- pMON73963 Figure 3
- Nostoc punctiforme Nostoc genomic DNA is used as a DNA template source for polymerase chain reaction amplification (PCR) of DNA for cloning into bacterial cells so as to express the Arabidopsis thaliana protein corresponding to SEQ ID NO: 2, SEQ ID NO: 4, or Nostoc protein corresponding to SEQ ID NO: 50.
- PCR polymerase chain reaction amplification
- thermocycler needed for performing PCR reaction was procured from Applied Biosystems (Perkin-Elmer Corp., Applied Biosystems Div., Foster City, CA).
- the polynucleotide sequence of the amplicon produced from Arabidopsis is shown in SEQ ID NO:l and SEQ ID NO: 3.
- PCR primer pairs SEQ ID NO: 57 and SEQ ID NO: 58 were used for performing the reaction to obtain the Arabidopsis amplicon.
- Arabidopsis amplicons were subcloned into a pET-28b vector (E coli expression vector, Novagen, Madison, WI, USA) and were sequenced by using sequencing primers SEQ ID NO: 59 and SEQ ID NO: 60 to confirm the sequence of the cloned polynucleotide molecule.
- Nostoc amplicons were generated by using Nostoc genomic DNA as template with primers pairs SEQ ID NO: 61 and SEQ ID NO: 63 or SEQ ID NO: 62 and SEQ ID NO: 63.
- Nostoc amplicons were also subcloned in to the pET-28b vector and were sequenced by using sequencing primers SEQ ID NO: 64 and SEQ ID NO: 65 to confirm the polynucleotide sequence of the cloned polynucleotide molecule.
- Primer Sequence for amplification and sequencing of molecules of invention from Arabidopsis and Nostoc are shown in Figure 10. These constructs contained a candidate from Arabidopsis SEQ ID NO: 3 and the closest Nostoc homologue SEQ ID NO: 49, both with and without an N-terminal His-tag. An N-terminal His-tag was chosen based on the crystal structures (Nakajima et al.
- Protein extracts were prepared by freeze thawing and sonication as described Cull and McHenry (Cull M and McHenry CS. Methods in Enzymology 182, 147-153, 1990). Pellets were resuspended in an appropriate volume of either 50 mM Tris-HCI pH 7.4 250 mM NaCI and 500 ⁇ M EDTA with 0.05% CHAPS and ImM ABESF or 50 mM KH 2 PO 4 pH-7.2 250 mM NaCI and 250 ⁇ M EDTA with 0.05% CHAPS and ImM ABESF, buffer.
- Protein concentrations were 1.5 to 1.7 mg /mL,based on the method of Bradford (Bradford M, Anal Biochem, 72, 248, 1976 and Bio-Rad Laboratories Procedure bulletin 1123) with BSA standard (Bradford reagent and BSA was from Bio-Rad Laboratories Headquarters 1000 Alfred Nobel Drive Hercules, CA 94547).
- the assays described below used 10 or 15 uL of this solution
- Spectrophotometric assays were used for determination of enzyme activities of purified proteins. Assays were based on earlier described methods (Portsteffen et al. Phyochemistry, 37, 391, 1994). Consumption or production of NADPH was observed at 340 nm. Assays were performed in a Varian Gary 50 Bio spectrophotometer (Varian Instruments inc. AA, ICP, UV, Fluorescence Products 679 Springvale RoadMulgrave, Victoria 3170Australia).
- NADPH solutions were freshly made and checked by UV-vis for degradation.
- Enzyme solutions were supplemented with ImM MgCl 2 and ImM Zn Cl 2 which were at 1.5 to 1.7 mg /mL concentrations.
- the assays used 10 or 15 ⁇ L of this solution (ca. 15 to 22 ⁇ g per reaction).
- Stock solutions of the substrates (20 to 500mM) were made up in a reaction buffer or as noted.
- Steroids solutions were made in 100% EtOH. From these stock solutions additions to the assays were made directly. Precipitation of steroids was observed.
- the soluble concentration of steroids in reaction mixture is not accurately known in each case.
- good estimate of substrate (sterol) concentration would be 100 to 200 ⁇ M for a saturated solution under these conditions.
- Table 8 Zero Order Result Table using acetone as substrate (60mM) shows evidence of reductase activity. Turnover (consumption of NADPH) is observed in the reductive direction. Slopes (for acetone, lines 1 and 2) indicate the difference from control is 10 fold. Controls (lines 3 and 4) were identical except for the omission of acetone.
- Start start time
- Stop stop time
- Slope change in absorbance over time
- a start is starting absorbance
- a stop is ending absorbance.
- Table 9 Zero Order Result Table. This table shows elected data for 5 ⁇ -androstane-3,17- dione in a reductase assay. This example shows the results for three reactions with acetone, three reactions with 5 ⁇ -androstane-3,17-dione and two controls. Table 8 shows the initial rates as estimated by linear fit between 0 and 5 minutes. The results for 5 ⁇ -androstane-3,17-dione shown here are the best of any substrate at the same concentration (approx 200 ⁇ M). 4- androstene-3,17-dione is the next best substrate with a rate on the order of 2.5 to 4 times slower than this under the same conditions (not shown). Acetone results at similar concentrations (Table 7 shows results at high acetone concentration) are on the order of 10 to 20 times slower and nearly at background levels. Similar results have been obtained with multiple batches of enzyme and substrate. Sample Start Stop Slope A A S.D.
- Start start time
- Stop stop time
- Slope change in absorbance ovei • time
- a start is starting absorbance
- a stop is ending absorbance
- RNA is purified from tissue using Trizol reagent from Life Technologies (Gibco BRL, Life Technologies, Gaithersburg, Maryland U.S.A.), as recommended by the manufacturer.
- Poly A+ RNA (mRNA) is purified using magnetic oligo dT beads essentially as recommended by the manufacturer (Dynabeads, Dynal Corporation, Lake Success, New York U.S.A.).
- the cDNA libraries are plated on LB agar containing the appropriate antibiotics for selection and incubated at 37° for sufficient time to allow the growth of individual colonies.
- the plasmid DNA is isolated from each clone using Qiaprep plasmid isolation kits, using the conditions recommended by the manufacturer (Qiagen Inc., Santa Clara, California U.S.A.).
- the template plasmid DNA clones are used for subsequent sequencing.
- a commercially available sequencing kit such as the ABI PRISM dRhodamine Terminator Cycle Sequencing Ready Reaction Kit with AmpliTaq® DNA Polymerase, FS, is used under the conditions recommended by the manufacturer (PE Applied Biosystems, Foster City, CA).
- the cDNAs of the present invention are generated by sequencing initiated from the 5' end or 3' end of each cDNA clone. Entire inserts or only part of the inserts (ESTs or expressed sequenced tags) are sequenced.
- a number of DNA sequencing techniques are known in the art, including fluorescence- based sequencing methodologies. These methods have the detection, automation and instrumentation capability necessary for the analysis of large volumes of sequence data.
- the 377 and 3700 DNA Sequencer Perkin-Elmer Co ⁇ ., Applied Biosystems Div., Foster City, CA
- fluorescent dye-labeled sequence reaction products are detected and data entered directly into the computer, producing a chromatogram that is subsequently viewed, stored, and analyzed using the corresponding software programs.
- These methods are known to those of skill in the art and have been described and reviewed (Birren et al, Genome Analysis: Analyzing DNA,1, Cold Spring Harbor, New York, the entirety of which is herein inco ⁇ orated by reference).
- the generated ESTs (including any full-length cDNA inserts or complete coding sequences) are combined with ESTs and full-length cDNA sequences in public databases such as GenBank. Duplicate sequences are removed, and duplicate sequence identification numbers are replaced. The combined dataset is then clustered and assembled using Pangea Systems tool identified as CAT v.3.2. First, the EST sequences are screened and filtered, e.g. high frequency words are masked to prevent spurious clustering; sequence common to known contaminants such as cloning bacteria are masked; high frequency repeated sequences and simple sequences are masked; unmasked sequences of less than 100 bp are eliminated.
- the screened and filtered ESTs are combined and subjected to a word-based clustering algorithm which calculates sequence pair distances based on word frequencies and uses a single linkage method to group like sequences into clusters of more than one sequence, as appropriate.
- Clustered sequences are assembled individually using an iterative method based on PHRAP/CRAW/MAP providing one or more self-consistent consensus sequences and inconsistent singleton sequences.
- the assembled clustered sequence files are checked for completeness and parsed to create data representing each consensus contiguous sequence (contig), the initial EST sequences, and the relative position of each EST in a respective contig.
- the sequence of the 5' most clone is identified from each contig.
- the initial sequences that are not included in a contig are separated out.
- SEQ ID NO: 1 exhibits 52.857% percent identity with its closest known functional gene, identified using the BLAST 2.2.1 software with BLOSUM62 matrix and "no Filter” options (Genbank accession number gi
Abstract
Description






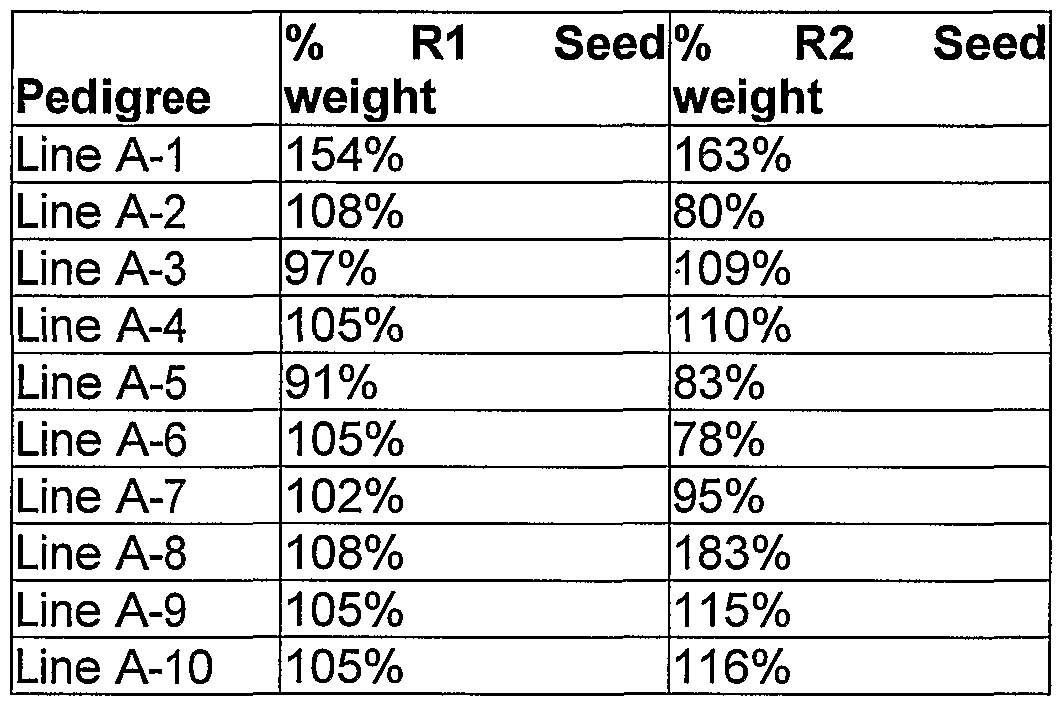






Claims
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2003237834A AU2003237834A1 (en) | 2002-05-15 | 2003-05-14 | Method of increasing plant organ and seed size in a plant |
| EP03736593A EP1504106B1 (en) | 2002-05-15 | 2003-05-14 | Method of increasing plant seed weight |
| DK03736593.9T DK1504106T3 (en) | 2002-05-15 | 2003-05-14 | Method of increasing the weight of a plant seed |
| US10/513,167 US7335812B2 (en) | 2002-05-15 | 2003-05-14 | Method of increasing plant organ and seed size in a plant |
| AT03736593T ATE473281T1 (en) | 2002-05-15 | 2003-05-14 | METHOD FOR INCREASE SEED WEIGHT IN A PLANT |
| DE60333280T DE60333280D1 (en) | 2002-05-15 | 2003-05-14 | PROCESS FOR INCREASING THE SEED WEIGHT IN A PLANT |
| US12/001,901 US7994402B2 (en) | 2002-05-15 | 2007-12-13 | Method of increasing plant organ and seed size in a plant |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US38110002P | 2002-05-15 | 2002-05-15 | |
| US60/381,100 | 2002-05-15 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10513167 A-371-Of-International | 2003-05-14 | ||
| US12/001,901 Division US7994402B2 (en) | 2002-05-15 | 2007-12-13 | Method of increasing plant organ and seed size in a plant |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2003096797A2 true WO2003096797A2 (en) | 2003-11-27 |
| WO2003096797A3 WO2003096797A3 (en) | 2004-11-18 |
Family
ID=29550070
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2003/014989 WO2003096797A2 (en) | 2002-05-15 | 2003-05-14 | Method of increasing plant organ and seed size in a plant |
Country Status (9)
| Country | Link |
|---|---|
| US (2) | US7335812B2 (en) |
| EP (1) | EP1504106B1 (en) |
| AR (2) | AR040020A1 (en) |
| AT (1) | ATE473281T1 (en) |
| AU (1) | AU2003237834A1 (en) |
| DE (1) | DE60333280D1 (en) |
| DK (1) | DK1504106T3 (en) |
| ES (1) | ES2346863T3 (en) |
| WO (1) | WO2003096797A2 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2353387A1 (en) | 2010-02-05 | 2011-08-10 | Bayer CropScience AG | Use of succinate dehydrogenase (SDH) inhibitors in the treatment of plant types in the sweet grass family |
| WO2014161908A1 (en) | 2013-04-05 | 2014-10-09 | Bayer Cropscience Nv | Brassica plants comprising mutant da1 alleles |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012112529A1 (en) | 2011-02-14 | 2012-08-23 | Xyleco, Inc. | Processing biomass |
| US9297021B2 (en) | 2011-11-02 | 2016-03-29 | University Of North Texas | MtNIP regulated plants with significantly increased size and biomass |
| JP2021501231A (en) | 2017-10-27 | 2021-01-14 | キシレコ インコーポレイテッド | Biomass processing method |
Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0050424A1 (en) | 1980-09-24 | 1982-04-28 | Cetus Corporation | Method for producing an analytical antibody probe, an analytical antibody probe, and a method for analysing a sample for certain antibodies |
| EP0084796A2 (en) | 1982-01-22 | 1983-08-03 | Cetus Corporation | HLA typing method and cDNA probes used therein |
| EP0120516A2 (en) | 1983-02-24 | 1984-10-03 | Rijksuniversiteit Leiden | A process for the incorporation of foreign DNA into the genome of dicotyledonous plants; Agrobacterium tumefaciens bacteria and a process for the production thereof |
| EP0154204A2 (en) | 1984-03-06 | 1985-09-11 | Mgi Pharma, Inc. | Herbicide resistance in plants |
| US4582788A (en) | 1982-01-22 | 1986-04-15 | Cetus Corporation | HLA typing method and cDNA probes used therein |
| EP0201184A2 (en) | 1985-03-28 | 1986-11-12 | F. Hoffmann-La Roche Ag | Process for amplifying nucleic acid sequences |
| US4683194A (en) | 1984-05-29 | 1987-07-28 | Cetus Corporation | Method for detection of polymorphic restriction sites and nucleic acid sequences |
| EP0237362A1 (en) | 1986-03-13 | 1987-09-16 | F. Hoffmann-La Roche Ag | Process for detecting specific nucleotide variations and genetic polymorphisms present in nucleic acids and kits therefor |
| EP0258017A2 (en) | 1986-08-22 | 1988-03-02 | F. Hoffmann-La Roche Ag | Purified thermostable enzyme and process for amplifying, detecting, and/or cloning nucleic acid sequences using said enzyme |
| US5107065A (en) | 1986-03-28 | 1992-04-21 | Calgene, Inc. | Anti-sense regulation of gene expression in plant cells |
| US5231020A (en) | 1989-03-30 | 1993-07-27 | Dna Plant Technology Corporation | Genetic engineering of novel plant phenotypes |
| US5641876A (en) | 1990-01-05 | 1997-06-24 | Cornell Research Foundation, Inc. | Rice actin gene and promoter |
| US5689042A (en) | 1995-03-29 | 1997-11-18 | Wisconsin Alumni Research Foundation | Transgenic plants with altered senescence characteristics |
| US5859347A (en) | 1992-03-19 | 1999-01-12 | Monsanto Company | Enhanced expression in plants |
| US6147278A (en) | 1985-10-25 | 2000-11-14 | Monsanto Company | Plant vectors |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE69816059T2 (en) * | 1997-02-10 | 2004-04-22 | National Research Council Of Canada, Ottawa | GENE THAT CODES FOR THE PYRUVATE DEHYDROGENASE KINASE OF PLANTS |
| CA2263067A1 (en) | 1999-02-26 | 2000-08-26 | The Australian National University | Method of modifying plant morphology, biochemistry and physiology |
| JP2003525051A (en) * | 2000-03-01 | 2003-08-26 | リサーチ・アンド・ディベロップメント・インスティチユート・インコーポレイテッド | Transgenic plants with improved seed yield, biomass, and harvest index |
| AU8681101A (en) * | 2000-08-24 | 2002-03-04 | Scripps Research Inst | Stress-regulated genes of plants, transgenic plants containing same, and methodsof use |
| AU2001289843A1 (en) * | 2001-08-28 | 2002-02-13 | Bayer Cropscience Ag | Polypeptides for identifying herbicidally active compounds |
| US20090183270A1 (en) * | 2002-10-02 | 2009-07-16 | Adams Thomas R | Transgenic plants with enhanced agronomic traits |
-
2003
- 2003-05-14 DE DE60333280T patent/DE60333280D1/en not_active Expired - Lifetime
- 2003-05-14 AT AT03736593T patent/ATE473281T1/en not_active IP Right Cessation
- 2003-05-14 EP EP03736593A patent/EP1504106B1/en not_active Expired - Lifetime
- 2003-05-14 AU AU2003237834A patent/AU2003237834A1/en not_active Abandoned
- 2003-05-14 WO PCT/US2003/014989 patent/WO2003096797A2/en not_active Application Discontinuation
- 2003-05-14 ES ES03736593T patent/ES2346863T3/en not_active Expired - Lifetime
- 2003-05-14 DK DK03736593.9T patent/DK1504106T3/en active
- 2003-05-14 US US10/513,167 patent/US7335812B2/en not_active Expired - Lifetime
- 2003-05-15 AR ARP030101701A patent/AR040020A1/en active IP Right Grant
-
2007
- 2007-12-13 US US12/001,901 patent/US7994402B2/en active Active
-
2009
- 2009-08-12 AR ARP090103126A patent/AR073039A2/en not_active Application Discontinuation
Patent Citations (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0050424A1 (en) | 1980-09-24 | 1982-04-28 | Cetus Corporation | Method for producing an analytical antibody probe, an analytical antibody probe, and a method for analysing a sample for certain antibodies |
| EP0084796A2 (en) | 1982-01-22 | 1983-08-03 | Cetus Corporation | HLA typing method and cDNA probes used therein |
| US4582788A (en) | 1982-01-22 | 1986-04-15 | Cetus Corporation | HLA typing method and cDNA probes used therein |
| EP0120516A2 (en) | 1983-02-24 | 1984-10-03 | Rijksuniversiteit Leiden | A process for the incorporation of foreign DNA into the genome of dicotyledonous plants; Agrobacterium tumefaciens bacteria and a process for the production thereof |
| EP0154204A2 (en) | 1984-03-06 | 1985-09-11 | Mgi Pharma, Inc. | Herbicide resistance in plants |
| US4683194A (en) | 1984-05-29 | 1987-07-28 | Cetus Corporation | Method for detection of polymorphic restriction sites and nucleic acid sequences |
| US4683202B1 (en) | 1985-03-28 | 1990-11-27 | Cetus Corp | |
| EP0201184A2 (en) | 1985-03-28 | 1986-11-12 | F. Hoffmann-La Roche Ag | Process for amplifying nucleic acid sequences |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US6147278A (en) | 1985-10-25 | 2000-11-14 | Monsanto Company | Plant vectors |
| EP0237362A1 (en) | 1986-03-13 | 1987-09-16 | F. Hoffmann-La Roche Ag | Process for detecting specific nucleotide variations and genetic polymorphisms present in nucleic acids and kits therefor |
| US5107065A (en) | 1986-03-28 | 1992-04-21 | Calgene, Inc. | Anti-sense regulation of gene expression in plant cells |
| EP0258017A2 (en) | 1986-08-22 | 1988-03-02 | F. Hoffmann-La Roche Ag | Purified thermostable enzyme and process for amplifying, detecting, and/or cloning nucleic acid sequences using said enzyme |
| US5231020A (en) | 1989-03-30 | 1993-07-27 | Dna Plant Technology Corporation | Genetic engineering of novel plant phenotypes |
| US5641876A (en) | 1990-01-05 | 1997-06-24 | Cornell Research Foundation, Inc. | Rice actin gene and promoter |
| US5859347A (en) | 1992-03-19 | 1999-01-12 | Monsanto Company | Enhanced expression in plants |
| US5689042A (en) | 1995-03-29 | 1997-11-18 | Wisconsin Alumni Research Foundation | Transgenic plants with altered senescence characteristics |
Non-Patent Citations (66)
| Title |
|---|
| "Current Protocols in Molecular Biology", 1989, JOHN WILEY & SONS, pages: 6.3.1 - 6.3.6 |
| "Fromm,.UCLA Symposium on Molecular Strategies for Crop Improvement", 1990, KEYSTONE |
| ALTSCHUL ET AL., JMOI. BIOL., vol. 215, 1993, pages 403 - 410 |
| ALTSCHUL, STEPHEN F. ET AL., NUCLEIC ACIDS RES., vol. 25, 1997, pages 3389 - 3402 |
| ALTSCHUL, STEPHEN F. ET AL.: "Nucleic Acids Res.", vol. 25, 1997, WEBB MILLER, AND DAVID J. LIPMAN, pages: 3389 - 3402 |
| AMMIRATO ET AL.: "Handbook of Plant Cell Culture - Crop Species", 1984, MACMILLAN PUBL. CO. |
| AUSUBEL ET AL.: "Current Protocols in Molecular Biology", 1989, JOHN WILEY & SONS |
| BACK ET AL., PLANT MOL. BIOL., vol. 17, 1991, pages 9 |
| BEVAN, NUCLEIC ACIDS RES., vol. 12, 1984, pages 8711 - 8721 |
| BI ET AL., PLANT J., vol. 8, 1995, pages 235 - 245 |
| BINDING: "Regeneration of Plants, Plant Protoplasts", 1985, CRC PRESS, pages: 21 - 73 |
| BUSK ET AL., PLANT J., vol. 11, 1997, pages 1285 - 1295 |
| CHEN ET AL., PLANT J., vol. 10, 1996, pages 955 - 966 |
| DATTA ET AL., BIO-TECHNOLOGY, vol. 8, 1990, pages 736 - 740 |
| DE VEYLDER ET AL., PLANT CELL |
| DELLAPORTA ET AL., STADLER SYMPOSIUM, vol. 11, 1988, pages 263 - 282 |
| EVANS ET AL.: "Handbook of Plant Cell Culture", 1983, MACMILLILAN PUBLISHING COMPANY, article "Protoplasts Isolation and Culture", pages: 124 - 176 |
| FRALEY ET AL., PROC. NATL. ACAD. SCI. USA, vol. 80, 1983, pages 4803 - 4807 |
| FROHMAN ET AL., PROC. NATL. ACAD. SCI. USA |
| GAN S.; AMASINO R.M., PLANT PHYSIOL., vol. 113, 1997, pages 313 - 319 |
| GAN S.; AMASINO R.M., SCIENCE, vol. 270, 1995, pages 1986 - 1988 |
| GAN; AMASINO, SCIENCE, vol. 270, 1995, pages 1986 - 1988 |
| GUARENTE L.; RUVKUN G.; AMASINO R.M., PROC. NATL ACAD. SCI USA, vol. 95, 1998, pages 1034 - 1036 |
| HAYASHIMOTO, PLANT PHYSIOL., vol. 93, 1990, pages 857 - 863 |
| HENSEL L.L ET AL., THE PLANT CELL, vol. 5, 1993, pages 553 - 564 |
| HERRERA-ESTRELLA ET AL., NATURE, vol. 303, 1983, pages 209 |
| HINCHEE ET AL., BIO/TECHNOLOGY, vol. 6, 1988, pages 915 - 922 |
| HUANG ET AL., PLANT MOL. BIOL., vol. 33, 1997, pages 125 - 139 |
| IKATU ET AL., BIO/TECHNOL., vol. 8, 1990, pages 241 - 2 42 |
| J. J. DOYLE; B. S. GAUT, PLANT MOLECULAR BIOLOGY, vol. 42, 2000, pages 1 - 23 |
| JEFFERSON ET AL., BIOCHEM. SOC. TRANS., vol. 15, 1987, pages 17 - 19 |
| JEFFERSON ET AL., EMBO J., vol. 6, 1987, pages 3901 - 3907 |
| JEFFERSON, PLANT MOL. BIOL, REP., vol. 5, 1987, pages 387 - 405 |
| K. WISING ET AL., ANN. REV. GENETICS, vol. 22, 1988, pages 421 |
| KATZ ET AL., J. GEN. MICROBIOL., vol. 129, 1983, pages 2703 - 2714 |
| KIRCH, PLANT MOL. BIOL., vol. 33, 1997, pages 897 - 909 |
| KLEE ET AL., ANN. REV. PLANT PHYS., vol. 38, 1987, pages 467 - 486 |
| KLEE ET AL., BIO-TECHNOLOGY, vol. 3, no. 7, 1985, pages 637 - 642 |
| LIU ET AL., PLANT PHYSIOL., vol. 115, 1997, pages 397 - 407 |
| LOHMAN K.M. ET AL., PHYSIOLOGIA PLANTARUM, vol. 92, 1994, pages 322 - 328 |
| MULLIS ET AL.: "ymp. Quant. Biol.", vol. 51, 1986, COLD SPRING HARBOR, pages: 263 - 273 |
| NEEDLEMAN; WUNSCH, J. MOL. BIOL., vol. 48, 1970, pages 443 - 453 |
| OW ET AL., SCIENCE, vol. 234, 1986, pages 856 - 859 |
| POTRYKUS ET AL., MOL. GEN. GENET., vol. 199, 1985, pages 183 - 188 |
| RATTAN ET AL.: "Protein Synthesis: Post-translational Modifications and Aging", ANN. N.Y ACAD SCI., vol. 663, 1992, pages 48 - 62 |
| S. HENIKOFF; J. G. HENIKOFF, PROC. NATL. ACAD. SCI. U.S.A., vol. 89, 1992, pages 10915 - 10919 |
| SAKAI ET AL., PLANT CELL PHYSIOL., vol. 37, 1996, pages 906 - 913 |
| SAMBROOK ET AL.: "Molecular Cloning - A Laboratory Manual", 1989, COLD SPRING HARBOR LABORATORY |
| SAMBROOK ET AL.: "Molecular Cloning, A Laboratory Manual", 1989, COLD SPRING HARBOR PRESS |
| SEIFTER ET AL., METH. ENZYMOL., vol. 182, 1990, pages 626 - M |
| SHEEN ET AL., SCIENCE, vol. 274, 1996, pages 1900 - 1902 |
| SHIMAMOTO ET AL.: "Nature", vol. 338, 1989, pages: 274 - 276 |
| STALKER ET AL., J. BIOL. CHEM., vol. 263, 1988, pages 6310 - 6314 |
| STREIT ET AL., MOL. PLANT MICROBE INTERACT., vol. 10, 1997, pages 933 - 937 |
| SUTCLIFFE ET AL., PROC. NATL. ACAD SCI., vol. 75, 1978, pages 3737 - 3741 |
| T. E. CREIGHTON; W. H. FREEMAN; COMPANY: "Proteins - Structure and Molecular Properties", 1993 |
| T. F. SMITH; M. S. WATERMAN, J. MOL.BIOL, vol. 147, 1981, pages 195 - 197 |
| THILLET ET AL., J. BIOL. CHEM., vol. 263, 1988, pages 12500 - 12508 |
| THOMAS H.; STODDART J. L., ANN. REV. PLANT PHYSIOL, vol. 31, 1980, pages 83 - 111 |
| UKNES ET AL., PLANT. CELL, vol. 5, 1993, pages 159 - 169 |
| VASIL ET AL., BIO/TECHNOLOGY, vol. 10, 1992, pages 667 - 674 |
| VASIL ET AL., BIO/TECHNOLOGY, vol. 8, 1990, pages 429 - 434 |
| W.R. PEARSON, GENOMICS, vol. 11, 1991, pages 635 - 650 |
| WEAVER L.M. ET AL., PLANT MOL. BIOL., 1998, pages 455 - 469 |
| WOLD, F.: "Post-translational Covalent Modification of Proteins", 1983, ACADEMIC PRESS, article "Post-translational Protein Modifications. Perspectives and Prospects", pages: 1 - 12 |
| ZUKOWSKY ET AL., PROC. NATL. ACAD SCI., vol. 80, 1983, pages 1101 - 1105 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2353387A1 (en) | 2010-02-05 | 2011-08-10 | Bayer CropScience AG | Use of succinate dehydrogenase (SDH) inhibitors in the treatment of plant types in the sweet grass family |
| WO2011095496A2 (en) | 2010-02-05 | 2011-08-11 | Bayer Cropscience Ag | Use of succinate dehydrogenase (sdh) inhibitors in treating plant species of the family of true grasses |
| WO2014161908A1 (en) | 2013-04-05 | 2014-10-09 | Bayer Cropscience Nv | Brassica plants comprising mutant da1 alleles |
| US10287603B2 (en) | 2013-04-05 | 2019-05-14 | Bayer Cropscience Nv | Brassica plants comprising mutant DA1 alleles |
Also Published As
| Publication number | Publication date |
|---|---|
| US20090133162A1 (en) | 2009-05-21 |
| EP1504106A2 (en) | 2005-02-09 |
| EP1504106B1 (en) | 2010-07-07 |
| EP1504106A4 (en) | 2006-06-21 |
| DK1504106T3 (en) | 2010-10-11 |
| AU2003237834A8 (en) | 2003-12-02 |
| AR040020A1 (en) | 2005-03-09 |
| ES2346863T3 (en) | 2010-10-21 |
| AR073039A2 (en) | 2010-10-06 |
| WO2003096797A3 (en) | 2004-11-18 |
| ATE473281T1 (en) | 2010-07-15 |
| US20060168693A1 (en) | 2006-07-27 |
| US7994402B2 (en) | 2011-08-09 |
| US7335812B2 (en) | 2008-02-26 |
| DE60333280D1 (en) | 2010-08-19 |
| AU2003237834A1 (en) | 2003-12-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6307126B1 (en) | Nucleic acid encoding GAI gene of Arabidopsis thaliana | |
| EP2241624A2 (en) | Nucleic acid molecules and their use in plant sterility | |
| US6781035B1 (en) | Leafy cotyledon1 genes and their uses | |
| PL189711B1 (en) | Method of improving seeds | |
| US20080209585A1 (en) | Genes which produce staygreen characteristics in maize and their uses | |
| US7994402B2 (en) | Method of increasing plant organ and seed size in a plant | |
| AU2001241600B2 (en) | Leafy cotyledon1 genes and their uses | |
| US7612258B2 (en) | Nucleic acid molecules associated with plant cell proliferation and growth and uses thereof | |
| US20100281580A1 (en) | Use of a gene encoding a histidine protein kinase to create drought resistant plants | |
| US20040006796A1 (en) | Methods of modulating cytokinin related processes in a plant using B3 domain proteins | |
| US7932434B2 (en) | Late blight resistance gene from wild potato | |
| EP1699926B1 (en) | Stress tolerant plants and methods thereof | |
| EP1377666A2 (en) | Plant growth regulating genes, proteins and uses thereof | |
| WO2005017114A2 (en) | Method of increasing metabolism and stress tolerance in a plant | |
| ZA200601187B (en) | Materials and methods for the modulation of cyclin-dependent kinase inhibitor-like polypeptides in maize | |
| AU2005253642B2 (en) | Nucleic acid molecules and their use in plant male sterility | |
| AU2002304741A1 (en) | Plant growth regulating genes, proteins and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PH PL PT RO RU SC SD SE SG SK SL TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2003736593 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2006168693 Country of ref document: US Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10513167 Country of ref document: US |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWP | Wipo information: published in national office |
Ref document number: 2003736593 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: JP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10513167 Country of ref document: US |